Received 10 October 1998 | Accepted 23 December 1998 | Published 31 December 1998
Reprint
Received 10 October 1998 | Accepted 23 December 1998 | Published 31 December 1998 |
Download Reprint |
Structure-Function Relationships in the Four Repeats of Human Interphotoreceptor Retinoid-Binding Protein (IRBP)
John M. Nickerson, Gui-Ru Li, Ze-Yu
Lin, Naoko Takizawa, Jing-Sheng Si, Eleanore A. Gross
Department of Ophthalmology, Emory Eye Center, Emory University, Atlanta GA 30322
Correspondence to: John M. Nickerson, Ph.D., Room B5602, Emory Eye Center, 1327 Clifton Road, N.E., Atlanta, GA, 30322, Phone: (404) 778-4411; FAX: (404) 778-3331; email: litjn@emory.edu
Abstract
Purpose: Interphotoreceptor retinoid-binding protein (IRBP) binds hydrophobic ligands in the interphotoreceptor space. Human IRBP consists of 1230 amino acids in four 300 amino acid long repeats. We asked: 1. Whether each of the four repeats can bind retinoids or fatty acids, 2. Whether each repeat can prevent retinol degradation in aqueous solutions, 3. Whether a ligand can stabilize the protein from thermal denaturation, 4. Whether the four repeats can be further classified into two groups. Our rationale was to make each repeat from the human cDNA and then examine structural and functional characteristics.
Methods: Individual repeats were produced in E. coli and the whole protein was expressed in baculovirus. Binding properties with all-trans-retinol were characterized by ligand fluorescence enhancement. The quenching of protein fluorescence by retinol, 9-cis-retinal, all-trans-retinoic acid, ß-ionine, [alpha]-ionine, trans-parinaric acid, and DHA was also examined. Binding curves were analyzed by nonlinear regression. Prevention of retinol decomposition was measured by absorption spectroscopy. Circular dichroism was examined in the far UV range to study protein secondary structure and the near UV range to study ligand binding effects on the tryptophan environment.
Results: Temperature dependent denaturation suggests that EcR1 is the most stable of the four repeats. Each repeat possesses the capability of binding 9-cis-retinal, all-trans-retinol, all-trans retinoic acid, docosahexaenoic acid, [alpha]- and ß-ionine, and trans-parinaric acid. Protein fluorescence quenching by retinol and retinol fluorescence enhancement assays yielded similar binding parameters for each repeat. Each expressed repeat prevents the degradation of retinol in aqueous solutions.
Conclusions: The data contrast with the idea that two or more repeats are needed to bind one molecule of ligand. Each repeat binds both retinoids and analogs, suggesting that each has multiple ligand binding sites or one binding site with affinity for different ligands. Together, the results suggest that each repeat retains all functions of the whole protein. However, there are distinguishing characteristics among the repeats in their ligand binding properties, though the four repeats cannot be classified into just two distinctive groups. Last, these data fit well with the current model of multiple binding sites in IRBP derived from quadruplication of an ancestral monomeric binding protein.
Introduction
Vitamin A (all-trans-retinol, retinol hereafter) is essential as a cofactor in the visual cycle. Deficiency of retinol leads to visual dysfunction, first manifested as night blindness. The metabolic processing of retinol in the eye has been studied extensively. The RPE is necessary for the regeneration of functional rhodopsin in the photoreceptors [1]. The RPE initially receives retinol from the bloodstream. In RPE, enzymes can esterify retinol and store it mainly as retinyl palmitate. Retinol isomerase is located uniquely in the RPE [2] establishing the need for the directional transport of retinoid isomers to and from the RPE and photoreceptor cell. Unbound retinoids are toxic, membranolytic, and labile. Thus, retinoids are thought to be transported while bound to specific soluble binding proteins [3].IRBP mediates retinoid transfer from the RPE cell to outer segments. For retinoids to reach the photoreceptor, they must first traverse the interphotoreceptor space (IPS), the space between the neural retina and the RPE [4,5]. Structure and order within the IPM change with illumination. More IRBP is concentrated near the apical and basal parts of the outer segments in light while it is more dispersed in the dark [6]. IRBP is the predominant soluble protein present in the monkey [7] and cow IPM [8].
Flannery et al [9] showed that retinol could be supplied to the apical face of RPE cells in culture and that later IRBP, but not other retinoid binding proteins, could facilitate the removal of 11-cis-retinal from the same face. Carlson and Bok [10] showed that tritiated retinol could be presented to the basolateral side of RPE cell cultures and later tritiated 11-cis-retinal could be recovered from the apical face via IRBP but not by other retinoid binding proteins. Okajima et al. [11,12] showed similar results in live RPE frog eyecup preparations: IRBP could mediate these processes while other retinoid binding proteins are much less effective. It remained to be shown whether intact RPE cells are required for IRBP to be able to retrieve 11-cis-retinal.
Adler and coworkers [13,14] recently pursued this important question, and they asked whether a subcellular fraction (microsomal membranes) of RPE cells can support the IRBP mediated removal of 11-cis-retinal from the RPE cell. They showed that microsomes can take up 3H-11-cis-retinal, and bovine IRBP can remove the 3H-11-cis-retinal from the microsomes. (No other subcellular or cellular machinery from the RPE cell is required). This demonstration shows that many subcellular components (lysosomes, nuclei, cytosolic proteins etc.) are not required for IRBP-assisted removal of 11-cis-retinal from the RPE cell. Correspondingly, this suggests that the extraction probably does not employ intracellular signaling pathways. In their experiments, liver microsomes substitute well for RPE microsomes. Thus, no specific proteins or even any protein in the microsomal membrane is required to mediate the process. They did not test whether protein free membranes or simple model membranes such as phosphatidyl choline (PC) single unilamellar vesicles (SUVs) could substitute for the microsomes. Nor did they test whether specific fragments of human IRBP could remove retinal from microsomes. Importantly, their work showed that IRBP could remove or extract 11-cis-retinal from the RPE membranes, but that other retinoid binding proteins (BSA and sRBP) were much less effective in this capacity.
Ho et al. [15] found that IRBP impedes the diffusion of all-trans-retinol through the aqueous environment between donor and acceptor small unilamellar vesicles (SUVs), suggesting a buffering or absorption function. IRBP protects retinoids from chemical degradation in aqueous environments up to or exceeding 48 hours in vitro. Without protection, substantial retinoid degradation occurs within one half hour at 37 °C [16].
Noy and coworkers found that docosahexaenoic acid (DHA) can profoundly affect the amount of retinoids binding to IRBP, and they suggested a model for the transport of 11-cis-retinal from RPE to the photoreceptor [17]. Because DHA is more concentrated in the photoreceptor than RPE membranes, and because DHA releases retinal from IRBP, the model is that IRBP can accept retinal near the RPE. Once close to the photoreceptor, IRBP releases retinal by competition with DHA. The DHA gradient thus contributes to the transfer of retinal from its production site to its point of use. Also according to this model, bovine IRBP possesses two kinds of binding sites for retinoids. One site is a hydrophobic or deep binding groove that can bind retinol and markedly enhance its fluorescence. The second site is a shallow site to which retinol can bind, but its fluorescence is not enhanced by binding to this site. Retinal can bind to both sites [18]. But only retinal bound to the second site (the shallow one) is regulated by DHA, which increases the off rate by 8-fold [17].
While these experiments suggest an important role for IRBP in the Vitamin A cycle, they have not revealed how substructures in IRBP carry out their functions or where the binding sites for retinoids exist in the protein. In most of these experiments the source of IRBP was cattle. It would be more beneficial from a medical viewpoint to study human IRBP instead. Our experiments employ human IRBP and its fragments for all experiments [19]. One basic question is whether each of the four repeats is a fully functional binding protein. Other possibilities include that each repeat retains only a subset of the intact protein's functions. Previously we showed that each of the four repeats in IRBP has binding capacity independent of the other repeats, and we found that none of the repeats is inactive. Here we confirm this prior observation and expand on that analysis. Also we begin to consider whether each repeat can fully carry out all of IRBP's properties: Does each repeat work as a miniature version of the full-size IRBP? Here we test the binding properties of the four individual repeats with several other analogs and retinoids. For example, we used [alpha]- and ß-ionine as models of retinoids lacking the polyene chain, and parinaric acid, a straight chain unsaturated compound containing four double bonds as a model lacking the cyclohexenyl ring. We used these as ligands to test the roles of the polyene chain and cyclohexenyl ring in binding to each repeat.We specifically compare results obtained by retinol fluorescence enhancement versus those from quenching of tryptophan (trp, W) fluorescence in the proteins. We assayed the binding of retinol to IRBP by varying the protein concentration at a constant retinol concentration. We also examined the tryptophan environment of each repeat by fluorescence and circular dichroism. Last, we examine the far UV region by CD to study how each repeat denatures with increasing temperature. All the data highlight the great similarity in functional characteristics of the four individual repeats, suggesting that each acts autonomously with many, if not all, of the functions of the intact whole protein.
Methods
Ligands
All-trans-retinol (70% pure, R-7632) and 9-cis-retinal (>95% pure, R-5754), all-trans-retinoic acid (>98% pure, R-2625) and docosahexaenoic acid (DHA) (approximately 99% pure, D-2534) were obtained from Sigma, St. Louis, MO. They were dissolved in 100% ethanol. The concentration was estimated using an extinction coefficient of 46,000 A/M/cm at 325 nm for retinol [20], 36,080 A/M/cm at 373 nm for 9-cis-retinal [21], and 45,000 A/M/cm at 350 nm for all-trans retinoic acid [20]. Trans-parinaric acid was obtained from Molecular Probes, Inc., Eugene, OR. It was dissolved in ethanol and concentration estimated via extinction coefficient of 85,000 A/M/cm at 298 nm, as given by the manufacturer. [alpha]-Ionine (approximately 90%, e = 14300 A/M/cm at 228.5 nm [22]) and ß-ionine (approximately 96%, e = 8700 A/M/cm at 293.5 nm [22]) were obtained from Aldrich (Milwaukee, WI). All ligands were prepared and used under subdued red or orange light.
Protein Production
Wild type (WT), EcR1, EcR2, EcR3, and EcR4 were produced in the baculovirus system and E. coli, and purified to near homogeneity, as described [19].
Circular Dichroism (CD) Measurements
CD spectroscopy was carried out as described [23,24] and [19], using a Jasco (Easton, MD) J-715 spectropolarimeter. Proteins were exchanged into 5 mM NaPO4, pH 6.5 by gel filtration with Sephadex G-25 columns (Pharmacia, Piscataway NJ) or centrifugal filtration (Centricon 30 or Microcon 10 filters, Amicon Inc, Beverly, MA). These two techniques served to remove Tris, NaCl, EDTA, and other absorbing substances, which interfere with CD readings below about 210 nm. Usually protein concentrations were in the 0.2 to 0.5 mg/ml range or as indicated in figure legends. Far UV CD spectra were collected in 0.5 mm pathlength cells from 280 to 180 nm, with a bandwidth of 1.0 nm, resolution of 0.1 nm, response of 4 seconds, and speed of 10 nm/min. For temperature ramp experiments, the samples heated at a rate of about 1 °C/min and spectra were collected at 5 °C intervals from 25 to 95 °C. The proteins were incubated for 2 min at each temperature before scanning to assure equilibrium was reached. For near UV experiments similar experimental conditions were employed except a 1 cm pathlength was used and the scan wavelength range was 450-250 nm. Protein concentrations for the near UV experiments were 4.5 µM and the ligand concentration was 9 µM for trans-parinaric acid. For the all-trans-retinol near UV experiments, the ligand concentration was 67 µM, and the protein concentrations were 67, 18, 29, and 6.7 µM for EcR1 to 4, respectively. These were the highest concentrations of the proteins available. CD spectra were truncated once the phototube voltage exceeded 800 volts to achieve adequate signal to noise ratios.
Fluorescence Measurements
Fluorescence measurements and binding curve titrations were performed using 1 µM solutions of wild type or variants of IRBP in 10 mM Tris pH 7.5, 2 mM EDTA, and 500 mM NaCl. 700 µl volumes were used in 1 cm excitation by 0.2 cm emission quartz cuvettes. Measurements were made at ambient room temperature, 23-24 °C, using a Spex Fluorolog FL2T2 photon counting spectrofluorometer (Instruments SA, Edison, NJ) and collected as photons counted per second (cps). Static measurements were integrated for 2 seconds. Wavelength scans were carried out at 1 nm/s. For titrations, 0.5 or 1 µl aliquots of the ligand dissolved in 100% ethanol were added to the cuvette and mixed by pipetting with a plastic transfer pipet. After 100 s, a measurement was made, exposing the sample to light for only about 5 s. From 14 to 22 additions of ligand were made, but at no time did the ethanol concentration exceed 3%, and usually the final concentration was 2 to 2.5%. For fluorescence enhancement experiments with all-trans-retinol, excitation was at 331 nm, and emission was at 479 nm. For all fluorescence quenching experiments excitation was at 280 nm and emission was at 336 nm.
Analysis of Binding Curves by Nonlinear Regression
We analyzed fluorescence enhancement data quantitatively as described by Baer et al. [25] and precisely as according to [19]. For fluorescence quenching, we modified the expression slightly as given below. Solvent effects, photobleaching, and ligand degradation during the titrations were not separately considered. The magnitude of the quenching (Qx) was calculated by subtracting the fluorescence (Fx) at a total ligand concentration, X, from the fluorescence at zero ligand concentration, Fo. Since buffer with the same specified total ligand concentration exhibits slight fluorescence, this value (buffx) was added. That is, Qx = (Fo - Fx) + buffx. The paired data (X, Qx) are then fitted to
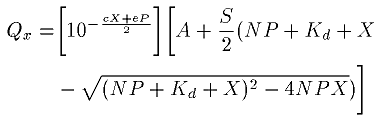
where:
Qx is the magnitude of the quenched fluorescence, the dependent variable, at a fixed total ligand concentration of X (measured in cps),
X is the total concentration of ligand in the cuvette and is the independent variable (measured in µM),
A is the difference in the background fluorescence between the two cuvettes, the one used for protein and the other used for the buffer blank (measured in cps),
S is the initial slope, that is, the change in fluorescence enhancement per unit change in total ligand concentration, at X=0 (in cps/µM),
c is an aggregate constant mostly the product of the extinction coefficients of ligand at the excitation and emission wavelengths and pathlengths (which result in the inner filter effect) plus a factor that compensates for photobleaching and damage to the ligands and protein during the assay (in A/µM),
Kd is the dissociation constant (in µM),
N is the number of independent ligand binding sites per polypeptide,
P is the protein concentration (in µM),
e is the molar extinction coefficient of the protein at the excitation wavelength (280 nm) multiplied by the excitation pathlength (in A/µM). The extinction coefficient was calculated from the exact amino acid sequences employing ProtParam at the ExPASy site following the method of Gill and von Hippel [26]. Protein absorption at the emission wavelength of 336 nm was assumed to be negligible and was ignored.
The expression, 10-((cX+eP)/2), compensates for sample absorbance causing the inner filter effect. Curve fitting was calculated by nonlinear regression employing SigmaPlot version 4.0 (Jandel Scientific, San Rafael, CA). Usually about 11-13 iterations were needed for convergence to a tolerance of 10-8.
Bmax is defined as the maximum amount of ligand that can be bound to a given amount of protein, as measured in units of fluorescence intensity. Bmax is calculated as the product of N and S from each individual titration curve.
Results & Discussion
Comparison of Retinol binding by tryptophan quenching
Each of the human repeats and WT intact human IRBP were tested for their ability to bind retinol and several other ligands by measuring the amount of quenching of fluorescence of tryptophan residues within the proteins. Tryptophan fluorescence is uniquely sensitive to the immediate surroundings and solvent environment of this amino acid. Even if a ligand absorbs no light emitted from tryptophan, the ligand can still profoundly affect and quench tryptophan fluorescence [27]. A ligand quenches fluorescence once it complexes or collides with an excited state tryptophan such that the latter returns to the ground state without emitting a photon. Figure 1 shows the fluorescence quenching of each individual repeat by retinol. The parameters from the curve fit are shown in Table 1.
Previously we showed that each repeat possesses a functional retinol binding site and 16 AP binding site by fluorescence enhancement, but the amount of enhancement varies among the repeats [19], with Repeats 1 and 2 enhancing fluorescence more than Repeats 3 and 4. This suggested the hypothesis that Repeats 1 and 2 correspond to Noy's Site 1, a deep hydrophobic site, [18,28] and Repeats 3 and 4 behave more like Noy's Site 2, a shallow site. Noy and coworkers [18,29] suggest that the second shallow site does not cause any retinol fluorescence enhancement, but their experiments may not totally exclude some fluorescence enhancement. Retinol bound to the protein solely via interaction at either end of the ligand should prevent translational motion of the ligand and might cause a slight enhancement of fluorescence.
To test whether each repeat binds retinol by an independent technique, we assayed for retinol binding by examining intrinsic trp fluorescence quenching. The results revealed that each repeat specifically binds retinol (Figure 1, Table 1). This result confirms the previous data obtained by fluorescence enhancement, but there is no pattern of Repeat 1 and 2 behaving as one binding class while repeats 3 and 4 act as a second kind. The results also imply that the microenvironments near the trps in each repeat are altered by binding. We also assayed several other ligands by trp quenching to test whether they too bind to each repeat. These additional assays show that each of the four repeats, by itself possesses functional binding sites for these several related small hydrophobic molecules. Each of the four different binding interactions is characterized as a high affinity and saturable event, and is therefore a specific binding process. Trp quenching results are tabulated in Table 1, Table 2, Table 3, Table 4, Table 5, and Table 6.
As shown in Table 1, each of the four repeats has a Kd for retinol in the 0.2 to 0.7 µM range, which is a little higher but not too different from the Kd found by retinol fluorescence enhancement for each repeat [19]. The relative binding capacities (Bmax) of the sum of the individual repeats is about 1.6 times the capacity in the intact wild type protein. The same ratio is found in our previous study with retinol fluorescence enhancement [19]. The number of binding sites per polypeptide ranges from a high of 1.73 in WT to a low of 0.25 in EcR3. Among the four repeats, there is no clustering of repeats into two distinctive classes based on Kd, N, and Bmax.
Regarding the curve-fitting process and the derived parameters, there is some degree of codependence of the S and N parameters, and EcR3 has the highest S parameter. This raises the possibility that a less likely binding curve might fit the data well, giving a higher N with a correspondingly lower S. This may also be indicated by the comparatively high CV associated with this S-value. The differences between the curve fit parameters found with intrinsic trp fluorescence quenching versus the values from the retinol fluorescence enhancement may reflect experimental error as the average CV is about 34%.
Other biologically relevant causes of the differences between the retinol binding properties measured by trp quenching and fluorescence enhancement may be that retinol fluorescence enhancement is an intimate measurement of the interaction of retinol within the binding site, whereas trp quenching is a measurement of any interaction within a larger radius of 5 nm from any of the four tryptophans per repeat. These interactions 1. may not be within the same binding site or domain in the protein, 2. may not reflect the same binding event within the same site (the ligand might rattle from one spot (which gives fluorescence enhancement) to another that generates trp quenching, all within a large binding site), or 3. may represent two or more binding sites measured by trp quenching and just one binding site giving retinol fluorescence enhancement, while the other gives no enhancement as in Noy and coworkers' two-site model. Further complications with the trp fluorescence measurements are that Repeats 1 and 2 possess four trps while Repeats 3 and 4 have just three trps, and these amino acids may not be located or oriented the same in the four different repeats. Similarly the trps might be slightly more or less buried relative to the surface of each repeat. Thus, one repeat might not quench as much as another despite possessing identical Kd and N. Thus, each repeat appears to bind one retinol molecule (but there are complications to the analysis).
Measurement of 9-cis-retinal binding to individual repeats
Table 2 and Figure 2 show the analysis of binding curves obtained by intrinsic tryptophan fluorescence quenching. These data show that each of the individual repeats possesses a high affinity and saturable binding site for 9 cis-retinal. The Kds are about 0.2 to 0.75 µM, in the physiological range. The Kds, Ns and Bmaxs all suggest that each repeat specifically binds 9-cis-retinal.
The numbers of binding sites for retinol (Table 1) and retinal (Table 2) for WT IRBP are about the same (1.6-1.7 sites) by trp fluorescence measurement. This number is the same as that determined by Chen et al. [18] for bovine IRBP (N =1.68) for retinol by trp fluorescence quenching. The Repeat 1 and 2 binding sites appear to have twice the Bmaxs of Repeats 3 and 4. Possibly, there are twice as many binding sites in the first two repeats, or the trps in Repeats 1 and 2 are positioned differently from those in Repeats 3 and 4, such that the former are quenched more readily than those in the latter repeats.
Comparison of DHA binding among the four repeats
Table 3 shows the curve fitting results from the binding curve analysis of DHA quenching of intrinsic tryptophan fluorescence. Kds from the four repeats are about the same ranging from about 0.2 to 0.4 µM. Each polypeptide appears to bind about 0.8 to 1.5 molecules of ligand and the binding capacities are about the same. The representative binding curves are shown in Figure 3. Why are the Bmaxs of the DHA curves so much lower than retinol or retinal? 1. DHA lacks the cyclohexenyl ring, and the pattern of double bonds is different suggesting that the excited state energy of the tryptophan may not be as readily dissipated by DHA. 2. DHA may not bind to the same site(s) as retinol or retinal, and consequently tryptophans may be farther away from DHA. 3. DHA may bind to Domain A, which lacks tryptophan, while retinol/al may bind to Domain B, which has three or four W's. The values of Kds for DHA compare favorably with the value (Ki = 0.78 µM) determined by competitive displacement of retinol [18]. The Bmaxs of about 1000 represent about 1/4 to 1/3 of the total trp fluorescence of the unquenched samples. While this fraction is not as great as in other cases, these fractions are about the same as observed by others for retinol quenching of intact bovine IRBP [18,29].
Binding curve analysis of [alpha]- and ß-ionine binding to each repeat
Table 4 and Figure 4 show the analysis and data from the quenching of intrinsic tryptophan fluorescence in the individual repeats. The Kds are about the same for each ligand in all the repeats, ranging from about 0.2 to 0.6 µM. However, there apparently are more binding sites for [alpha]-ionine than ß-ionine, suggesting that [alpha]-ionine may bind roughly two molecules per repeat versus about one ß-ionine per repeat. Bmax and S are lower than the values found for the binding of retinal or retinol suggesting that the polyene chain may be close to a least one tryptophan such that it can affect quenching of this amino acid. The polyene chain is known to play an important role in the fluorescence of retinol and presumably fluorescence enhancement. However, because the binding affinity of ionine is only a little lower than for retinol or retinal, it suggests that the polyene chain does not necessarily play a great role in the affinity of these ligands for the individual repeat. None of the results with [alpha]- or ß-ionine suggest that any of the repeats can be grouped into two distinct classes: The properties are basically the same among all four repeats.
The binding of all-trans-retinoic acid to IRBP
In Table 5, the binding of all-trans-retinoic acid to each repeat is examined. The Kds range from 0.5 to 0.75 µM, roughly the same as retinal and a little lower affinity than retinol. Each repeat binds roughly one retinoic acid molecule per polypeptide. These results are similar to the Ki (1.54 µM) [18] found by displacement of retinol from WT intact bovine IRBP. Again, we cannot cluster the four repeats into two distinctive classes as the binding parameters are all very similar among the four repeats.
The binding of trans-parinaric acid to the IRBP repeat
In Table 6 and Figure 5, the binding of trans-parinaric acid is considered. The Kds for each repeat are all quite low suggesting a tight binding interaction; however, the number of binding sites is very low, but consistent among the four repeats, with N averaging only about 0.17 binding sites per repeat. This suggests three principal alternatives: 1. The ligand takes on a shape that fits into the binding site only about 17% of the time. 2. The protein takes on a shape that can bind the ligand only about 17% of time (trivially, perhaps only 17% of the protein is native). 3. An allosteric affecter is missing, such that if it were present, the binding capacity would then approach N = 1. Additional less likely alternatives include: 1. Though optimal curve-fitting suggests low N's and high S's, the reciprocal nature of S and N might suggest that the S's are unusually high. Lower S's would yield higher N's. The S's for trans-parinaric acid seem higher than most of the other ligands. 2. High affinity, low capacity saturable binding could be mistaken for a small amount of fluorescence quenching from derived from changes in light scattering, solvent viscosity, increased sample turbidity, or adsorption. But quenching due to nonspecific binding of the ligand, solvent, or contaminants should not affect the analyses unless quenching is nonlinear, with large effects on quenching at low concentrations and smaller increments in quenching at high concentrations. This should not normally occur.
The analysis of the parinaric acid binding curve displays a component of the signal that represents a saturable amount of quenching, the Bmax, which is a fraction of the unquenched trp fluorescence large enough to suggest that the binding of parinaric acid to each repeat is a saturable process. Among the four repeats, the binding properties with trans-parinaric acid are about the same suggesting no clustering of the four repeats into the two hypothesized groups.
The high affinities in Table 6 are reminiscent of the binding of some fatty acid analogs, which all have very low Kds (15-120 nM) as assessed by fluorescence enhancement measurements [19,30]. This may suggest that the fatty acid analogs, DHA, and trans-parinaric acid may all bind to a similar or the same binding site, while the cyclohexenyl ring structure is binding either to a different binding site or a distinctly different environment within the same binding site. The results with these fatty acid binding studies differ somewhat from the results of Chen and coworkers [28] who found higher Kds by the competitive displacement of retinol from bovine IRBP. The tightest interaction was found with DHA at 0.78 µM, and palmitic acid has a Kd of 4.15 µM by their approach. The several-fold differences in the Kds suggest that the displacement assay may be measuring an interaction at one site, while the trp quenching and fluorescence enhancement assays may be measuring binding at an entirely different site. Regardless, it seems clear that fatty acids and analogs specifically bind to each of the four repeats.
Reverse titration of the fluorescence enhancement assay
Figure 6 shows a titration of protein added in increments to a constant concentration of retinol (1 µM) as the ligand. As expected, the results of this assay show that the same Kd is obtained irrespective of which agent serves as titrant, retinol or the protein (EcR2).
Circular Dichroism of IRBP and expressed repeats
CD has been used to characterize the gross shape of the four repeats previously [19]. We have broadened the scope of the CD studies in two areas: 1. CD spectral analysis in the near UV can be used to study the binding interaction of a ligand and a receptor. 2. Thermal denaturation plus CD in the far UV can be used to assess the stability of the repeats and might reveal two different classes secondary or tertiary structure among the four repeats. Regarding the former approach, via near-UV experiments, we find that normally achiral ligands may take on some chiral characteristics, probably by the introduction of a twist, when bound to each of the four repeats. The most distinctive effects on the CD signals of the ligand-repeat complex are shown in Figure 7A and Figure 7B. These figures show the spectra obtained from EcR1 and EcR3 and trans-parinaric acid as receptors and ligand. While there is little CD signal derived from only ligand or only protein, when combined, the CD signal shows clear maxima at about 280, 303, and 320, and distinct minima at about 295, 310, and 325. The magnitudes of the peaks and valleys differ between Repeats 1 and 3, however. These results suggest that the tryptophan and tyrosine microenvironment changes shape and the ligand changes optical properties upon binding. The comparative lack of CD signals in the complexes of EcR2 and EcR4 with trans-parinaric acid (data not shown) suggest that these two repeats form one class while EcR1 and EcR3 form a second class. However, near UV CD data (not shown) with retinol as the ligand suggest that Repeats 1 and 2 have similar CD signals and form a class while Repeats 3 and 4 form a second class. Thus, the trp microenvironments do not form a consistent pattern allowing us to clearly differentiate the four repeats into two different classes.
Thermal denaturation was used to assess the general stability of the repeats, which may suggest differences in structure among the four repeats. The stability of the protein in the presence and absence of ligand can help to appraise binding site characteristics of the protein in the far UV. The results of thermal denaturation and examination of CD in the far UV range are shown in Figure 8 and Table 7. The most striking transition is found in EcR1 where two families of CD spectral curves are observed. One family of curves, corresponding to spectra collected at temperatures of 25 to 55 °C, has a minimum at 208 nm of about -40 to -44 mDeg while the other family of curves, corresponding to temperatures of 70 to 95 °C, has a minimum of about -27 mDeg. A sharp transition, the melting temperature, which is typified by the greatest vertical spacing between two adjacent spectra, occurs between spectra collected at 60 and 65 °C. The residual CD signal at the highest temperature, 95 °C, probably reflects beta sheet structure, which is the most thermally stable secondary structure. Each repeat exhibits the same residual beta sheet. The melting temperature among the four repeats varies from a high of 62 to a low of 47 without ligand, suggesting that Repeat 1 is the most stable protein of the four. The addition of ligands to Repeats 1, 3, and 4 seems to have little effect on the stability of the proteins. This suggests that the ligand binding pocket contributes little to the inherent stability of the folded protein. But in EcR2 the presence of retinoic acid reduces stability. This suggests that the carboxyl head group of this ligand is close to or within the binding pocket where the carboxyl moiety might destabilize the folded structure, while Repeat 1-bound retinoic acid may be positioned differently.
Comparison of Intact WT IRBP with a mixture of the four repeats
Each single repeat contains a minimum unit that binds retinoids and fatty acids. Whether there are cooperative interactions among the four repeats in intact IRBP is the next question. If each repeat is an independent and fully functional unit, then a mixture of the four individual repeats should behave similarly to the intact WT IRBP. Because of the low concentration of the protein (1 µM) of each repeat and 1 µM intact WT IRBP, we do not anticipate collisional interaction among the four repeats, unless there is a very high affinity of one repeat for the next. Thus, deviations of the mix versus the intact protein should reflect positive or negative cooperativity within intact WT IRBP. Table 8 shows the Kds and number of binding sites for intact protein versus the equimolar mixture of the four repeats. These were obtained by fluorescence enhancement (Figure 9) and trp quenching (Figure 10) methods using all-trans-retinol as the ligand. We observe only small differences between the mix and intact protein in the number of binding sites. The Kd of the intact protein is lower than the Kd of the mixture by both the fluorescence enhancement and trp quenching methods (while there are no significant differences between the quenching and enhancement methods, ANOVA, post hoc Student-Newman-Kuels test, p<0.05). The number of binding sites in intact WT IRBP is higher than previous measurements (2.7 and 2.1 binding sites here versus 1.3 [19]), and the number of binding sites in the mix of four repeats is even higher (2.9 and 2.7). The latter numbers are very close to the previous sum of the number of binding sites of individually measured repeats (2.78, [19]). The trend appears that the intact protein binds with a little higher affinity, but with less binding capacity than the mixture of the four repeats. This may suggest cooperative interactions in the intact molecule.
Retinol Degradation Protection
Regarding IRBP's role in protection of retinol from degradation, we found that each of the four individual repeats can protect retinol from degradation. This was assayed at both 25 and 37 °C with approximately 4 µM protein and 8 µM all-trans retinol. The results from the 25 °C experiments are shown in Figure 11. Retinol degrades rapidly with time in the absence of protein, but with protein little obvious degradation could be detected by absorbance wavelength scans every few minutes for about two hours. EcR2 appears to protect retinol the best, while the effectiveness of the three other repeats is about the same as bovine serum albumin. Very similar results were obtained at 37 °C (data not shown). These results are consistent with results carried out with full length bovine IRBP [15,16], and suggest that each repeat functions by itself to protect all-trans-retinol from degradation.
Extraction of 9-cis-retinal from SUVs by EcR2
Our next objective was to consider whether a single repeat is capable of binding retinal derived from a membrane. Although the precise biological function of IRBP is not known, it seems likely that IRBP is unique among retinoid binding proteins in the capability of removing retinal from RPE apical membranes. To assay for the putative extractive or removal function that is uniquely associated with IRBP, we sought to devise a simple and rapid assay: Can EcR2 accept ligand donated from an artificial membrane vesicle? An equilibrium experiment suggested that this single repeat can accumulate retinal derived from PC small unilamellar vesicles (SUVs). PC SUVs were prepared containing 5 mol% 9-cis-retinal by the ethanol injection method [31]. These were mixed with Repeat 2, and we asked whether the 9-cis-retinal from the SUVs could bind to the protein and quench the intrinsic fluorescence of tryptophan within the protein (excitation at 280 nm, emission at 336 nm, using a 1 by 0.2 cm cuvette). Without addition of the SUVs, the Repeat 2 signal was 65200±5240 cps. After the addition of the retinal-SUVs the signal was 57270±1440 cps. The SUVs contributed a signal of 115±5 cps. SUVs with retinal gave 86±15 cps. Repeat 2 incubated with SUVs lacking retinal gave a signal of 65800±4130 cps. The errors represent the standard deviation, and all values take into consideration a two-fold dilution. The experiment was done three times. The protein was quenched significantly (ANOVA, post hoc Student-Newman-Kuels test, p<0.05) only in the presence of the retinal-SUVs. Control SUVs lacking the 9-cis-retinal but prepared the same way showed no quenching. The membrane system is a simple PC SUV suggesting that only this phospholipid is required to form a substrate for retinal extraction. This equilibrium experiment indicates that the 9-cis-retinal from the SUVs was transferred to the individual repeat. The mechanism of transfer (aqueous diffusion or collisional transfer) was not assessed, but the experiment shows that EcR2 serves as an acceptor for 9-cis-retinal donated from PC SUVs.
Kinetic analysis of the transfer of retinal from SUV to protein
The magnitude of the change and the small CV of the above experiment suggested that we could carry out a kinetic analysis of the rate of transfer of retinal from the SUVs to IRBP. We mixed equal volumes of retinal-loaded SUVs and each repeat and measured the drop in tryptophan fluorescence in the protein as it accepted the ligand. The experiment employed the stopped flow instrumentation described in the Methods section, with a 0.3 by 0.3 cm pathlength cuvette (hence the raw fluorescence numbers cannot be directly compared to the above equilibrium experiment). The kinetics are shown in Figure 12. Curve-fitting with a single exponential (Figure 12A) yields residuals that are not randomly distributed about zero with time. However, curve-fitting with a double exponential function (Figure 12B) shows a significantly better fit by F-Test (p<1 x 10-14). The two time constants are 1.17 s-1 and 0.0574 s-1. About 40% of the protein's binding capacity is employed in the rapid transfer process while the remaining 60% is involved with the slower process. Future kinetic analyses will examine if the transfer reaction is first or second order, which might imply a diffusion model or contact-dependent mechanism. The existence of two rate constants might suggest easy-rapid access to one type of site and slower access and binding to a different class of binding site within a single repeat, as suggested [28] for intact bovine IRBP. Alternatively, we speculate that the two rate constants might reflect a mixed transfer mechanism, IE, a combination of aqueous transfer and collisional transfer mechanisms operating simultaneously. This would be strikingly different from the established aqueous transfer mechanism of all-trans-retinol transferring from vesicle to vesicle or from IRBP to a vesicle, as already established [15]. Another possible interpretation is that there are two off rates for 9-cis retinal leaving the SUV. This might occur if there are two kinds of SUVs within the SUV preparation or if retinal leaves the SUV in two different chemical states, such as a single molecule versus a cluster of molecules (for example, a tiny micelle). Another interpretation is that the slow rate represents degradation due to photobleaching or other chemical phenomena of an artifactual nature.
Experiments with the other three repeats showed the same basic behavior (Table 9). All showed a fast rate with a sub-second half-time and a slow rate with a half-time in the 10-15 second range. All showed a fast component of about 40-60% of the amount of the transfer-binding outcome. All four repeats possess the capability to accept 9-cis-retinal starting originally from SUVs, with no appreciable difference in the fast off rate from the SUVs among Repeats 2, 3, and 4. The exception is Repeat 1. It accepted retinal much faster than the other three repeats. This experiment was duplicated with different preparations of EcR1 and retinal-SUVs. This unusual difference in behavior of Repeat 1 versus the other three suggests a fundamentally different mechanism in binding this ligand. While we have not tested different concentrations of the acceptor, we speculate that there may be contact mediated transfer between EcR1 and SUVs that does not exist in the other three repeats.
The role of IRBP in vision
We now know that the IRBP protein itself is important for sight, since homozygous knockout mice exhibit reduced ERGs and short outer segments [32]. However, the precise function of IRBP that is apparently so essential remains unknown: Is it the 11-cis-retinal binding property or is it something else?
The results from ligand quenching of intrinsic tryptophan fluorescence (1) support a central hypothesis that IRBP contains multiple binding sites with at least one site per repeat, (2) suggest an essential role for IRBP in the visual process, and (3) suggest a role for IRBP and retinoids in preventing retinal degeneration. First, multiple binding sites are indicated by the capacity of each repeat to bind ligands such as retinol, 9-cis-retinal, retinoic acid, and [alpha]- and ß-ionine. The number of binding sites is generally near one per repeat. This conclusion is supported by both the fluorescence enhancement assay methods and intrinsic protein fluorescence quenching data. Second, the essential role of IRBP in the visual process is proven by the knockout mouse and supported by the unique presence of IRBP in the IPS and the binding capability for retinoids as shown here and previously. Third, the abnormal function and morphology of the photoreceptors in the knockout mouse, retinal degeneration in avitaminosis, slight improvements or decrease in the rates of deterioration in RP patients upon vitamin A palmitate therapy, and the facilitation of extraction of 11-cis-retinal all suggest that IRBP plays a role in preventing retinal degeneration.
Fluorescence enhancement studies versus trp quenching results
Previous studies showed that Repeats 1 and 2 have significantly more fluorescence enhancement of retinol than Repeats 3 and 4. These results can be explained by two models. Model 1 is that Repeats 1 and 2 each possess two retinol binding sites while Repeats 3 and 4 each have just one. In this model, each binding site exhibits about the same amount of fluorescence enhancement. Model 2 is that Repeats 1 and 2 each possess one binding site that has twice the fluorescence enhancement as the single site in each of Repeats 3 and 4. Our trp quenching data suggest that the repeats have equal binding capacity for retinol, suggesting that Model 2 holds. These results, suggesting just one (or at least equal numbers) retinol binding site per repeat, are supported by the trp quenching studies with other ligands where there are no major differences among the four repeats in the number of binding sites for any ligand.
Examination of a shallow binding Site and a Deep binding Pocket.
Noy and coworkers have found evidence for two kinds of binding sites in bovine IRBP. Also, they measured the binding capacity of intact bovine IRBP for retinol by several approaches including 3H-retinol binding, and they conclude that there are two sites per polypeptide. Do Repeats 1 and 2 behave like Noy's Site 2, and do Repeats 3 and 4 behave like Site 1? We have tested several new nonfluorescent ligands for binding activity by CD and quenching of protein fluorescence spectroscopy. From these studies, we conclude that there are few major differences among the four repeats in ligand binding and other tested properties, suggesting that the four repeats cannot be clustered into two different groups, one possessing just a deep hydrophobic pocket and the other having just a shallow binding site.
DHA has a competitive effect on the binding of 11-cis-retinal to Noy's Site 2 but no effect on the binding of 11-cis-retinal to Site 1. This observation suggests that DHA might bind to Site 2 with high affinity, but only with low affinity to Site 1. Thus, the hypothesis that Repeats 1 and 2 may be Site 1-like and Repeats 3 and 4 may be Site 2-like, suggests Kds of about 1 µM with Repeat 3 or 4 and 10 µM or greater with Repeats 1 and 2. The binding properties (see Table 3 and Figure 3) measured by quenching of protein fluorescence, did not show these differences and in fact suggests that all four repeats bind DHA with a Kd of about 0.2 to 0.3 µM, and each repeat may possess a site of a type similar to the Site 2 described by Noy and colleagues. With CD, we expect to find DHA binding only to the shallow site. This should not involve large changes in the protein structure and only minimal changes to the protein's CD in the tryptophan range, and this is what we found (data not shown).
Noy and coworkers suggested that retinol binds to Site 1 deeply placed into a binding site (Site 1-like). They also suggest that only the cyclohexenyl ring binds tightly to Site 2. These properties suggest that [alpha]-ionine might bind to Site 2 but not necessarily to Site 1: [alpha]-ionine might not fit properly into the deep hydrophobic pocket of Site 1, but it might bind tightly to the shallow site of Site 2 by hydrophobic interactions as proposed for the binding of the cyclohexenyl ring of retinol [29]. On the other hand, ß-ionine might bind well to both Site 1 and Site 2. A ligand lacking the ß-ionine ring or with a broken cyclohexenyl ring might bind well to Site 1, but not to Site 2. The remaining parts of the ring and the polyene chain might adapt well to the deep binding groove of Site 1, but the residual parts of the ring and the polyene chain might no longer match Site 2. Thus ligands such as trans-parinaric acid might bind to Site 1 and not Site 2. In the future, it may be possible to employ other retinoid analogs that are less lipophilic or contain bulky groups that will not fit into the deep hydrophobic pocket of Site 1. These might provide a clear discrimination between Sites 1 and 2. Via protein fluorescence quenching measurements, we found that both [alpha]- and ß-ionine and parinaric acid bind to each of the four repeats. These results do not suggest that Repeats 1 and 2 possess only a site similar to the bovine Site 1 or that Repeats 3 and 4 only possess a site resembling the bovine Site 2. The data speculatively could be interpreted to suggest that each repeat contain one site similar to Site 1 and one similar to Site 2 described in bovine IRBP by Noy and colleagues. These two sites might even reside within a single large cavity that could accommodate two ligands simultaneously or in which a ligand might shift position or potentially "rattle" within the cavity.
On the other hand, the fluorescence enhancement data support the hypothesis that binding sites in Repeats 1 and 2 are deep hydrophobic pockets, and the binding sites in Repeats 3 and 4 are not as hydrophobic or confining. Alternative explanations include that Repeats 1 and 2 contain four trps and Repeats 3 and 4 contain only three trps. The differences in the number and position of the trps may profoundly affect the amounts of fluorescence enhancement or quenching despite very little difference in the affinity of retinol for or the capacity of the ligand binding sites. Nonetheless, the high affinities of retinoids (including retinol, 9-cis-retinal and all-trans-retinoic acid) and the analogs of the polyene chain (parinaric acid and DHA) and the cyclohexenyl ring ([alpha]- and ß-ionine) all suggest that each repeat is fully capable of binding retinoids.
Recent perceptive studies of the fourth module of Xenopus IRBP provide interesting data supporting many of the conclusions we draw in this paper [33]. Baer and coworkers find that this protein, corresponding to human Repeat 4, possesses the capability to bind roughly one retinol per polypeptide. A central fragment of Module 4 also retains high capacity and affinity to bind retinol while a fragment from the N-terminal end lacks binding capacity. The C-terminal end retains some binding capacity, suggesting a second binding site within a single repeat. These data may lend support to a model in which there are two different kinds of retinoid binding sites in each repeat (or module). In addition, the consistency of binding properties between human Repeat 4 and Xenopus Module 4 and the sequence conservation highlight the apparent importance of IRBP in the function of the visual system.
We have answered the central structure-function question of this paper, which was: Given the four-repeat structure of the amino acid sequence, can each repeat bind a ligand? We find that, indeed, each individual repeat binds retinoids and fatty acids and these ligand binding interactions are specific. We also find that the differences among the repeats in their ligand-binding properties seem far smaller than their similarities.
Lastly, our equilibrium and kinetic analyses suggest that a single repeat can function to accept retinal donated from a model membrane. Repeat 1 seems to be especially efficient in this property. This implies that IRBP can extract retinal from RPE membranes independent of other biological organelles, enzymes, and proteins in the lipid bilayer, and independent of an energy source.
Acknowledgements
Supported by NIH R01 EY10553, P01 EY06360, T32 EY07092, and an unrestricted grant from RPB to the Department of Ophthalmology, Emory University. Parts of this study were reported at ARVO, Ft. Lauderdale FL in May of 1998 [34].
References
1. Wald G. The molecular basis of visual excitation. Nature 1968;
219:800-7. ![]()
2. Bernstein PS, Law WC, Rando RR. Isomerization of
all-trans-retinoids to 11-cis-retinoids in vitro. Proc Natl Acad Sci U S
A 1987; 84:1849-53. ![]()
3. Chader GJ, Wiggert B, Lai YL, Lee L, Fletcher RT. Interphotoreceptor retinol-binding protein: a possible role in retinoid transport to the retina. Prog Retin Eye Res 1983; 2:163-89.
4. Feeney L. The interphotoreceptor space. II. Histochemistry of the
matrix. Dev Biol 1973; 32:115-28. ![]()
5. Feeney L. The interphotoreceptor space. I. Postnatal ontogeny in
mice and rats. Dev Biol 1973; 32:101-14. ![]()
6. Uehara F, Matthes MT, Yasumura D, LaVail MM. Light-evoked changes
in the interphotoreceptor matrix. Science 1990; 248:1633-6.
![]()
7. Pfeffer B, Wiggert B, Lee L, Zonnenberg B, Newsome D, Chader G.
The presence of a soluble interphotoreceptor retinol-binding protein
(IRBP) in the retinal interphotoreceptor space. J Cell Physiol 1983;
117:333-41. ![]()
8. Adler AJ, Martin KJ. Retinol-binding proteins in bovine
interphotoreceptor matrix. Biochem Biophys Res Commun 1982; 108:1601-8.
![]()
9. Flannery JG, O'Day W, Pfeffer BA, Horwitz J, Bok D. Uptake,
processing and release of retinoids by cultured human retinal pigment
epithelium. Exp Eye Res 1990; 51:717-28. ![]()
10. Carlson A, Bok D. Promotion of the release of 11-cis-retinal from
cultured retinal pigment epithelium by interphotoreceptor
retinoid-binding protein. Biochemistry 1992; 31:9056-62.
![]()
11. Okajima TI, Pepperberg DR, Ripps H, Wiggert B, Chader GJ.
Interphotoreceptor retinoid-binding protein: role in delivery of retinol
to the pigment epithelium. Exp Eye Res 1989; 49:629-44.
![]()
12. Okajima TI, Pepperberg DR, Ripps H, Wiggert B, Chader GJ.
Interphotoreceptor retinoid-binding protein promotes rhodopsin
regeneration in toad photoreceptors. Proc Natl Acad Sci U S A 1990;
87:6907-11. ![]()
13. Adler AJ, Edwards RB. IRBP uniquely removes 11-cis-retinal from isolated RPE membranes. Invest Ophthalmol Vis Sci 1995; 36:S6.
14. Adler AJ, Southwick RE. Binding of 11-cis retinoids to IRBP. Invest Ophthalmol Vis Sci 1998; 39:S422.
15. Ho MT, Massey JB, Pownall HJ, Anderson RE, Hollyfield JG.
Mechanism of vitamin A movement between rod outer segments,
interphotoreceptor retinoid-binding protein, and liposomes. J Biol Chem
1989; 264:928-35. ![]()
16. Crouch RK, Hazard ES, Lind T, Wiggert B, Chader G, Corson DW.
Interphotoreceptor retinoid-binding protein and alpha-tocopherol
preserve the isomeric and oxidation state of retinol. Photochem
Photobiol 1992; 56:251-5. ![]()
17. Chen Y, Houghton LA, Brenna JT, Noy N. Docosahexaenoic acid
modulates the interactions of the interphotoreptor retinoid-binding
protein with 11-cis-retinal. J Biol Chem 1996; 271:20507-15.
![]()
18. Chen Y, Saari JC, Noy N. Interactions of all-trans-retinol and
long-chain fatty acids with interphotoreceptor retinoid-binding protein.
Biochemistry 1993; 32:11311-8. ![]()
19. Lin ZY, Li GR, Takizawa N, Si JS, Gross EA, Richardson K,
Nickerson JM. Structure-function relationships in interphotoreceptor
retinoid-binding protein (IRBP). Mol Vis 1997; 3:17 <http://www.molvis.org/molvis/v3/p17/>.
![]()
20. Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding affinities of
retinol and related compounds to retinol binding proteins. Eur J Biochem
1976; 65:71-8. ![]()
21. Hubbard R, Brown PK, Bownds D. Methodology of vitamin A and visual pigments. Methods Enzymol 1971:18; 615-53.
22. Windolz M, Budavari S, Blumetti RF, Otterbein ES, editors. The Merck index: an encyclopedia of chemicals, drugs, and biologicals. 10th ed. Rahway (NJ): Merck; 1983.
23. Woody RW. Circular dichroism. Methods Enzymol 1995; 246:34-71.
![]()
24. Johnson WC Jr. Protein secondary structure and circular
dichroism: a practical guide. Proteins 1990; 7:205-14. ![]()
25. Baer CA, Kittredge KL, Klinger AL, Briercheck DM, Braiman MS,
Gonzalez-Fernandez F. Expression and characterization of the fourth
repeat of Xenopus interphotoreceptor retinoid-binding protein in E.
coli. Curr Eye Res 1994; 13:391-400. ![]()
26. Gill SC, von Hippel PH. Calculation of protein extinction
coefficients from amino acid sequence data. Anal Biochem 1989;
182:319-26. ![]()
27. Lakowicz JR. Principles of fluorescence spectroscopy. New York: Plenum Press; 1983.
28. Chen Y, Noy N. Retinoid specificity of interphotoreceptor
retinoid-binding protein. Biochemistry 1994; 33:10658-65.
![]()
29. Tschanz CL, Noy N. Binding of retinol in both retinoid-binding
sites of interphotoreceptor retinoid-binding protein (IRBP) is
stabilized mainly by hydrophobic interactions. J Biol Chem 1997;
272:30201-7. ![]()
30. Nickerson JM, Mody V, DeGuzman C, Heron KA, Marciante K, Boatright J, Si JS, Lin ZY. Ligand-binding properties of recombinant human IRBP. In: Anderson RE, LaVail MM, Hollyfield JG, editors. Degenerative diseases of the retina. New York: Plenum Press; 1995. p. 395-402.
31. Kremer JM, Esker MW, Pathmamanoharan C, Wiersema PH. Vesicles of
variable diameter prepared by a modified injection method. Biochemistry
1977; 16:3932-5. ![]()
32. Liou GI, Fei Y, Peachey NS, Matragoon S, Wei S, Blaner WS, Wang
Y, Liu C, Gottesman ME, Ripps H. Early onset photoreceptor abnormalities
induced by targeted disruption of the interphotoreceptor
retinoid-binding protein gene. J Neurosci 1998; 18:4511-20.
![]()
33. Baer CA, Retief JD, Van Niel E, Braiman MS, Gonzalez-Fernandez F.
Soluble expression in E. coli of a functional interphotoreceptor
retinoid-binding protein module fused to thioredoxin: correlation of
vitamin A binding regions with conserved domains of C-terminal
processing proteases. Exp Eye Res 1998; 66:249-62. ![]()
34. Nickerson JM, Lin ZY, Li GR, Takizawa N, Si JS, Gross EA. Structure-function relationships in human interphotoreceptor retinoid-binding protein (IRBP). Invest Ophthalmol Vis Sci 1998; 39:S422.