Received 26 August 1997 | Accepted 28 October 1997 | Published 30 December 1997
Reprint
Received 26 August 1997 | Accepted 28 October 1997 | Published 30 December 1997 |
Download Reprint |
Structure-function relationships in interphotoreceptor retinoid-binding protein (IRBP)
Ze-Yu Lin, Gui-Ru Li, Naoko Takizawa, Jing-Sheng Si, Eleanore A.
Gross, Kevin Richardson, John M. Nickerson
Department of Ophthalmology, Emory University, Atlanta, GA
Correspondence to: John M. Nickerson, Ph.D., Room B5602, Emory Eye Center, 1327 Clifton Road, N.E., Atlanta, GA, 30322, Phone: (404) 778-4411, Fax: (404) 778-3331, Email: litjn@emory.edu
Abstract
Purpose: Interphotoreceptor retinoid binding protein (IRBP) binds hydrophobic ligands in the retina. The polypeptide consists of 1230 amino acids in four 300 amino acid long repeats. We asked whether each of the four repeats can bind one retinoid or fatty acid analog. Our rationale was to make protein variants from the human cDNA bearing one or more of the repeats and examine binding capacities and dissociation constants.
Methods: Proteins were characterized by SDS-PAGE, western blotting, N-terminal sequencing, and CD spectroscopy. Binding properties with all-trans-retinol and 16-anthryloxy-palmitic acid (16-AP) were characterized by ligand fluorescence enhancement and curve fitting.
Results: Binding capacities varied according to the length of each protein. Each repeat possesses the capability of binding retinol and 16-AP.
Conclusions: The data contrast with the idea that two or more repeats are needed to bind one molecule of ligand. Each repeat binds a retinoid and fatty acid analog, suggesting that each has multiple ligand binding sites or one binding site with affinity for different ligands. Last, these data fit well with the current model of multiple binding sites in IRBP derived from quadruplication of an ancestral monomeric binding protein.
Introduction
Interphotoreceptor retinoid binding protein (IRBP) is found in the vertebrate eye in a space between the photoreceptor cells and the retinal pigment epithelium (RPE) (1). IRBP is unique to photosensitive tissues, that is, the retina and the pineal gland. Its apparent function is to bind and transport 11-cis-retinal from stores in the RPE to the photoreceptors. IRBP returns the spent form of vitamin A, all-trans-retinol, to the RPE where it is oxidized and isomerized to 11-cis-retinal again. IRBP aids in rhodopsin regeneration in vitro, and in all-trans-retinol uptake in vitro in RPE cells (2) and explant eyecups (3). Additionally, IRBP protects retinoids from chemical degradation in vitro (4). IRBP selectively extracts only 11-cis-retinal from RPE membranes (5). Ho et al. (6) suggest a buffering function. The existence of the visual cycle (Vitamin A cycle) and biochemical properties provide compelling arguments that IRBP should be necessary for sight (1, 7, 8). This role may not be unique. In chickens, purpurin is found in the IPS, and prostaglandin D synthase is found in the IPS of the rat retina (9). The synthase binds retinoic acid and retinal but, intriguingly, not all-trans-retinol (10). However, the unique presence of IRBP as the only retinoid binding protein of consequence in the monkey interphotoreceptor space (IPS) (11), its clear role in facilitating the directional uptake of 11-cis-retinal into rod outer segments (ROS) (12), and its roles in the recovery of rhodopsin sensitivity (13) and uptake of all-trans-retinol into retinyl ester stores in the RPE (3) demonstrate strong support for an essential role for IRBP in vision. For a detailed review of IRBP physiology, see (14).
IRBP is well conserved among the vertebrates and has been used to clarify issues in mammalian phylogeny (15, 16). IRBP bears similarities to proteins widely dispersed in nature (17, 18). While IRBP apparently exhibits no similarities to other retinoid binding proteins, a statistically significant match exists between Tsp (also known as prc) and IRBP (19). Whether this represents convergence or an ancestral relationship cannot be established by a single pairwise alignment. However, the detection of other similar proteins throughout the plant, animal and bacterial kingdoms and conservation of key residues in multiple sequence alignments suggest an ancient evolutionary relationship among these proteins. The conservation also implies that the tertiary shape has been useful for binding hydrophobic ligands such as peptides/proteins, fatty acids and retinoids (18, 20). This implies divergence from a common ancestor and homology among these proteins.
The structure of the IRBP gene and the protein is shown in Figure 1. There are two significant differences between IRBP and the other family members. First, almost all IRBP proteins contain four repeats, whereas the other known family members contain only one IRBP-like motif of about 220 amino acids in size (Domain B in Figure 1). Each repeat in IRBP is about 300 amino acids long (21-23). The second major difference is that the other members of the family possess an N-terminal extension of roughly 100 to 300 amino acids that makes up an additional domain unrelated to IRBP. We refer to the first 80 amino acids of the IRBP Repeat as Domain A. In IRBP these first 80 amino acids are homologous among IRBP Repeats. That is, within IRBP, Domain A from one repeat is homologous to Domain A from any other IRBP repeat. No homology exists between Domain A of IRBP and other N-terminal extensions preceding Domain B of other proteins in the family. Because Domain B has catalytic or binding functions in other proteins in the family, we hypothesize that each of the four repeats in IRBP has binding capacity independent of the other repeats.
To test this hypothesis, we created three sets of altered IRBP proteins: 1. A nested deletion set in which proteins were truncated at the C-terminus (named R123, R12+, R12-, and R1, from longest to shortest), 2. Point mutations in the IRBP protein (named G719S and R725C), and 3. Individual repeats cloned and expressed separately (named EcR1, EcR2, EcR3, and EcR4). These variants are defined in Table Ia. We compared these proteins among themselves and to wild type human IRBP by their capacity to bind ligands. We find that each separate repeat possesses a binding site for retinol and an analog of palmitic acid, experimentally verifying our hypothesis. Parts of this work were presented in abstract form (20, 24, 25) at annual meetings of the Association for Research in Vision and Ophthalmology from 1995-97.
Methods
Ligands
Retinol (70% pure, R-7632) was obtained from Sigma, St. Louis, MO. It was dissolved in 100% ethanol. The concentration was estimated using an extinction coefficient of 46,000 A/M/cm at 325 nm. 16-anthryloxy-palmitic acid (16-AP) was obtained from Molecular Probes, Inc., Eugene, OR. It was dissolved in ethanol and its concentration estimated via its extinction coefficient of 8200 A/M/cm at 361 nm.
Antibodies, SDS-PAGE, Protein Concentrations, and Western Blots
Western blots were performed as described (26) using a monoclonal anti-human IRBP, H3B5, generously provided by L. Donoso, Wills Eye Hospital, Philadelphia, PA. After SDS-PAGE, we electrophoretically transferred proteins to nitrocellulose membranes. The antibody (27), was diluted 1:1600 before use. Protein levels of IRBP and the constructed proteins were determined by A280 measurements in 6M guanidine (28) and using extinction coefficients calculated for each protein via ExPASy (http://expasy.hcuge.ch/www/tools.html). A polyclonal antibody was prepared in rabbits against an oligopeptide (amino acids 837-861) from Repeat 3 after cross-linking to keyhole limpet hemocyanin. The polyclonal antiserum was diluted 600-fold before use.
Clone Preparation
Standard recombinant DNA methods were used to construct, propagate, and isolate plasmids, and to sequence DNA and analyze the clones. Transplacement plasmids were constructed using pVL1392 (29) as the base vector, inserting cDNA fragments from pVL4200 (30) of the human IRBP cDNA (23) into the former vector. After ligation, the plasmids were transformed into competent SURE cells (Stratagene, La Jolla, CA) or JM109 (Promega, Madison, WI). The strategy was to include the signal peptide of IRBP so that the polypeptide would be translocated into the rER of the insect cells and later the protein would be secreted into the medium. As before, we used High FiveTM cells (InVitrogen Corp., San Diego, CA) and Excell 400 medium (JRH Biosciences, Lenexa, KS) with 0.5% fetal bovine serum for the expression and production of IRBP (30, 31).
Table Ib and Table Ic show how the protein-producing DNA constructs were prepared. As an example, the construct, pR1, is described in full below. Each other construct was prepared similarly. PCR products were subcloned into the identified vectors. pR1 contains amino acids -22 to 299 and was created by inserting a PCR fragment of pVL4200 from positions 4181 to 5336 into pVL1392. The primers included a NotI site in the forward primer (Primer 1) and an XbaI site and included two stop codons in the reverse primer (Primer 2). Table Ib shows the sequences of the primers used for this and other constructs. The digested PCR fragment was subcloned into pVL1392 cut with the same two enzymes. Ligations for this and succeeding constructs were performed by mixing a three-fold molar excess of insert with about 20 ng of vector. The reaction included 1 unit of T4 DNA ligase (GIBCO BRL, Gaithersburg, MD) and the manufacturer's ATP-containing buffer. After incubation for 2 h at 16 °C, approximately 20 ng of the recombinant vector was mixed with competent cells and transformed under conditions suggested by the manufacturer. Clones were recovered the next day. Minipreps (Wizard, Promega) of the colonies demonstrated inserts. Sequencing verified that the proper sites were reconstructed and that no errors were incorporated into any insert, except R12+ as discussed below. pR1 contains some IRBP 5' untranslated region. The produced protein was named R1. All the other constructs were created similarly and Table Ic shows details of how they were made. PCR products for clones pR1, pR12-, pR12+, and pR123 were cloned into pVL1392. PCR fragments for pG719S and pR725C were subcloned into pVL4200, from which the corresponding wild type restriction fragment had been excised, for baculovirus/insect cell protein expression.
The remaining four PCR products were cloned into pLEX (InVitrogen, La Jolla, CA) for protein expression in E. coli. For expression in E. coli, several vector-derived amino acids were left at the N-terminus and a poly(His) tail was included at the C-terminal end. The expressed proteins contained six histidines at the C-terminus, so that any proteolyzed or abnormal translation products would not likely contain the histidine tail and not bind to metal affinity columns during protein purification.
pR12+ is an aberrant clone but provides useful information. It resulted from the artifactual ligation of base 6373 from pVL4200 joined to the 3'-half NotI site of the pVL1392 vector. This suggests that the vector was not fully digested with Xbal and that the PCR fragment of this clone was digested with contaminating exo- and endonucleases, leaving base 6373 at the 3' terminus. Abnormal fill-ins and ligation appear to have yielded this clone. The resulting construct was normal at the N-terminal end but imperfect at the C-terminal end of the deduced protein: It possessed 29 non-IRBP amino acids at that end. The extra 29 amino acids originate from the residual multiple cloning site and the AcNPV polyhedrin gene fragment found in pVL1392. Despite the erroneous C-terminal end, the expressed protein still possesses important structural and functional characteristics. The produced protein was designated R12+.
Expression Conditions
Recombinant baculovirus were prepared as described in the manual provided by the kit manufacturer (MaxBac, version 1.6, InVitrogen, San Diego, CA). BaculoGold viral DNA (Pharmingen Inc., San Diego, CA) was used instead of wild type linearized AcNPV. Conditions for expression were described previously (30). Expression of the individual repeats in E. coli was carried out as described in the manufacturer's manual (Invitrogen) with minor modification. Typically, bacteria containing the expression plasmids were grown to an A550 of 0.5 at 30 °C. Tryptophan was added to a final concentration of 0.2 mg/ml and the cells were shaken at 225 rpm at 37 °C for four hours before harvesting by centrifugation.
Protein Purification
A previously described protocol was used to purify wild type recombinant human IRBP (30), and this method also was used to purify R1, G719S, and R725C. R12+ was purified using QAE ion exchange chromatography (30). Because R12- and R123 remain intracellular, they were purified from the insect cells, which were isolated by centrifugation at 2000 x g for 10 min. Typically 12 ml of cell pellet were homogenized in 40 ml of homogenization buffer (0.5% (V/V) Triton X-100 in 50 mM Tris pH 7.5 and 2 mM EDTA). The homogenate was centrifuged at 28,000 rpm in an SW30 rotor (Beckman, Schaumberg, IL) at approximately 120,000 x g for 30 min. The resulting pellet was extracted twice more in 40 ml of homogenization buffer. The final pellet (about 4 to 5 ml in volume) was homogenized in 90 ml of S Buffer (10 mM Tris pH 7.5, 100 mM NaCl, 2 mM EDTA) including 8 M urea. After centrifugation at 28,000 rpm for 30 min, the resulting supernatant was diluted to 150 ml final volume in S Buffer including 8 M urea and dialyzed against three changes of 6 liters of S Buffer. After dialysis, residual debris was removed by centrifugation at 10,000 X g for 10 min. The protein was concentrated by QAE chromatography on (Q-Sepharose Fast Flow resin, Pharmacia, Piscataway, NJ). The column was developed with an 18 ml 0.1 to 0.5 M NaCl gradient over 3.5 hours. R12- and R123 eluted at 0.35 to 0.4 M NaCl.
To purify E. coli expressed proteins, the cells were digested with 1 mg/ml lysozyme for 30 min on ice, sonicated for 1 min three times, and treated with 50 µg/ml RNase and 4.55 µg/ml DNase for 20 min on ice in 5 ml per gram of bacterial pellet in H Buffer (20 mM NaPO4, pH 7.4, 10 mM imidazole, 500 mM NaCl). Cell debris was removed by centrifugation at 100,000 x g for 20 min. For E. coli expressed Repeats 1, 3, and 4 (EcR1, EcR3, and EcR4), the proteins were readily soluble and were purified to virtual homogeneity by nickel nitrilotriacetic acid affinity chromatography: After loading the column (HisTrap, 1 ml bed volume, Pharmacia) with the protein containing solution, it was washed with 15-30 ml of H Buffer and protein eluted with 500 mM imidazole, 20 mM NaPO4, and 500 mM NaCl, pH 7.4. Most of the eluted protein usually appeared in the third and fourth fraction, where the volume of each fraction was 0.7 ml. This was dialyzed against 4 liters of S Buffer and subjected to QAE chromatography as above. For EcR2, which accumulated as inclusion bodies, cells were lysed as above, the debris was centrifuged, and the pellet extracted with 8 M urea in H Buffer by homogenization with the B pestle of a Dounce tissue grinder. The mixture was clarified by centrifugation (120,000 x g for 30 min), filtered through a Millex HV 0.45 µ low protein binding filter (Millipore), and the resulting solution was passed through a nickel nitrilotriacetic acid column, exceeding the column's capacity to bind His-tagged proteins. The polyhistidine tag allowed the recovery of the EcR2 protein in the denatured state. After first washing with H Buffer including 8 M urea to remove weakly bound his-rich proteins, EcR2 protein was eluted with 500 mM imidazole, 20 mM NaPO4, 500 mM NaCl, and 8M urea, pH 7.4. The protein was renatured by dialysis overnight against two changes of S Buffer. For dialysis, the protein concentration was adjusted below about 3 µM to minimize aggregation (32).
To evaluate the purity of the proteins, samples were subjected to SDS-PAGE. Gels were stained with Coomassie R-250 and analyzed by densitometric scanning on a 12-bit grayscale flatbed scanner and Molecular Analyst software (Bio-Rad, Hercules, CA). Dirt and specks on the gel images were removed by using a 7 x 7 noise reduction filter in this program. Peak areas were integrated after subtracting background and expressed as a percentage of total.
N-terminal Sequence Analysis and Numbering of the IRBP Sequences
According to the cDNA and genomic sequences the first initiator methionine in the IRBP reading frame occurs at amino acid position -22. The IRBP signal or pre-peptide consists of amino acids -22 to -6. The putative propeptide occurs at positions -5 to -1, and mature processed IRBP starts at positions -5 and +1, with about 50% starting at each position when protein is isolated from the IPS. This nomenclature is shown in Figure 2. Protein, separated by SDS-PAGE, was electrophoretically transferred to the PVDF membrane (33). After staining the membrane with Ponceau Red, the band was cut out. N-terminal sequence analysis was carried out on a gas phase sequenator using protein from solution or coupled to PVDF membrane as described (34).
Circular Dichroism (CD) Measurements
CD spectroscopy was carried out as described (35-37), using a Jasco (Easton, MD) J-715 spectropolarimeter. Proteins were exchanged into 5 mM NaPO4, pH 6.5 by gel filtration with Sephadex G-25 columns (Pharmacia) or centrifugal filtration (Centricon 30 or Microcon 10 filters, Amicon Inc, Beverly, MA). These two techniques served to remove Tris, NaCl, EDTA, and other absorbing substances, which interfere with CD readings below about 210 nm. CD spectra were collected in 0.5 mm pathlength cells from 350 to 170 nm, with a bandwidth of 1.0 nm, resolution of 0.1 nm, response of 4 seconds, and speed of 10-50 nm/min. Spectra were truncated once the phototube voltage exceeded 800 volts, which occurred at about 178 nm. [Absorbance measurements were simultaneously collected with the CD spectra and used to approximate the protein concentration by assuming an extinction coefficient of 31 A205 per mg/ml per cm]. After subtracting CD background, high frequency noise in the signals was reduced by using a Fourier transform in the instrument's software. Spectra were analyzed with several programs, including Contin (38), K2D (39), Selcon (40), and Varslc (41), to estimate secondary structural content, that is, [alpha]-helix, parallel and antiparallel ß -sheet, turns and other structures. Tertiary structure predicted from CD measurements was considered by using the program def_class.exe (42).
Fluorescence Measurements
Fluorescence measurements were performed using 1 µM solutions of wild type or variants of IRBP in 10 mM Tris pH 7.5, 2 mM EDTA, and 500 mM NaCl. 700 µl volumes were used in 1 cm pathlength quartz cuvettes. Measurements were made at ambient room temperature, 23-24 °C, using a Spex Fluorolog FL2T2 photon counting spectrofluorometer (Instruments SA, Edison, NJ) and collected as photons counted per second (cps). Static measurements were integrated for 2 seconds. Wavelength scans were carried out at 1 nm/sec. For titrations, 0.5 or 1 µl aliquots of the ligand dissolved in 100% ethanol were added to the cuvette and mixed by pipetting with a plastic transfer pipet. After 100 s, a measurement was made, exposing the sample to light for only about 5 s. From 14 to 22 additions of ligand were made, but at no time did the ethanol concentration exceed 3%, and usually the final concentration was 2 to 2.5%.
Analysis of Binding Curves by Nonlinear Regression
We analyzed the data quantitatively as described by Baer et al. (32), by curve fitting to the following expression, which is a slight variation of their formula. A 1 cm pathlength cuvette was used. Solvent effects, photobleaching, and ligand degradation during the titrations were not separately considered.
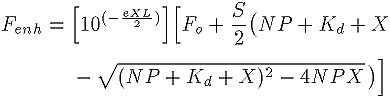
where:
Fenh is the fluorescence enhancement, that is, the dependent variable, and the difference between fluorescence of two cuvettes, one with protein and one without, at a fixed total ligand concentration. (measured in cps)
X is the total concentration of ligand in the cuvette and is the independent variable (measured in µM)
Fo is the offset of the background fluorescence between the two cuvettes, one with protein and the other with just buffer at zero ligand concentration (measured in cps)
S is the initial slope, that is, the change in fluorescence enhancement per unit change in total ligand concentration, at X=0 (in cps/µM)
e is the sum of the extinction coefficients of ligand at the excitation and emission wavelengths (in A/µM/cm)
Kd is the dissociation constant (in µM)
N is the number of independent ligand binding sites per polypeptide
P is the protein concentration (in µM)
L is the pathlength of the cuvette, which was 1 cm for both the excitation and emission paths.
Curve fitting was calculated by nonlinear regression employing SigmaPlot (Jandel Scientific, San Rafael, CA). Usually no more than 30 iterations were needed for convergence to occur with a tolerance of 10-9.
Here we define Bmax is the maximum amount of ligand that can be bound to a given amount of protein, as measured in units of fluorescence intensity. Bmax is calculated as the product of N and S.
Results
Protein Production and Purification
IRBP mutations (G719S and R725C) and nested deletions (R123, R12+, R12-, and R1) were introduced into baculovirus and used to infect large stocks of insect cells. Generally, the designed protein produced in the insect cells was secreted into the medium and was glycosylated. The proteins were successfully purified by Con A chromatography because all the variant proteins contained at least one complex carbohydrate NXT/S attachment site in their sequences (see Figure 1). While the proteins exhibited similar behavior to wild type IRBP during purification, especially ion exchange chromatography, two of the proteins, R123 and R12-, remained mainly in the insect cells, although minor amounts were secreted. Figure 3 shows the results of purification of several IRBP deletion proteins. The individual repeats, EcR1, EcR2, EcR3, and EcR4 were made in E. coli. EcR1, 3, and 4 were produced in soluble form and were purified to virtual homogeneity by chromatography on a nickel nitrilotriacetic acid affinity column and an anion exchange column. The other protein, EcR2, became insoluble in E. coli, but it too could be purified and renatured. The purified E. coli-produced proteins are shown in Figure 3b. To summarize, adequate amounts of highly purified proteins were obtained using the baculovirus and E. coli expression systems.
Western Blots and SDS-PAGE Analysis
The baculovirus-expressed proteins vary in purity by staining with Coomassie R-250 (Figure 3a), but are all >57% pure. The major bands constitute 100% of total for wild type and R725C, 84% for R1, 76% for G719S, 58% for R12-, 71% for R12+ (sum of the three bands), and 70% for R123. The E. coli-expressed individual repeats (Figure 3b) are almost 100% pure by staining of the SDS gel. Western blotting shows that the major bands all represent IRBP, as they all react with a monoclonal antibody (H3B5) or a polyclonal serum directed against an IRBP oligopeptide by immunostaining (Figure 3a and Figure 3b). EcR1 and EcR2 reacted strongly with H3B5 and showed a heavy brown reaction product (Figure 3b). Lighter but distinctly visible brown reaction products were observed with EcR3 and EcR4. EcR1, EcR3, and EcR4 all reacted with the polyclonal antibody made against amino acids 837-861. Experiments with pre-immune and nonimmune sera, sera against other antigens, and omission of primary sera showed no immunoreactions (data not shown).
To summarize, excepting R12+, there is an IRBP immunoreactive band of the anticipated size suggesting that each protein is produced correctly. The cause of the nonhomogeneity of R12+ is considered below. There are some differently sized molecular weight bands but these are generally minor constituents. These results validate the use of these proteins for further experimental work.
N-Terminal Amino Acid Sequencing
Sequence analysis of the first few amino acids established that the major protein in each preparation is the authentic and expected product. The absence of any secondary sequences shows the homogeneity of each single band seen on SDS PAGE and establishes the purity of the preparations. The sequences are shown in Figure 2. All begin with the GPTHLFQPSL... sequence as predicted from the gene and cDNA and prior sequencing at the amino acid level, except R1 and the E. coli expressed proteins. R1 begins with MREWLLVLLM..., which is the authentic IRBP signal sequence. Thus, N-terminal sequencing confirms that this protein encodes IRBP, but the signal sequence has not been removed. Figure 2 also shows that EcR1, EcR2, EcR3, and EcR4 begin with short leaders derived from the multiple cloning sites of the pLex vector, but the authentic sequences representing IRBP begin exactly as expected from conceptual translation of the DNA constructs. These data further verify that the purified proteins represent IRBP.
For R12+, the three major bands were cut out individually and analyzed. All three have the same N-terminal sequence suggesting that C-terminal truncation is occurring and that proteolytic processing happens by recognition of distinct sites in the protein. The C-terminal 29 amino acids of R12+ are not IRBP, suggesting that the processing is an artifact. However, even the shortest R12+ polypeptide is slightly larger than R12-. This defines a boundary on the location of C-terminal sites of R12+ protein processing. The same three bands in R12+ appeared in several different preparations. This repeated finding suggests that this proteolysis is a rapid intracellular event and possibly the consequence of a lack of folding of the artifactual sequence.
CD Spectra
To establish whether the variant proteins have folded into the same general conformation as wild type IRBP, we carried out circular dichroism studies. Hazard et al. (37) in a preliminary report examined the CD characteristics of recombinant human IRBP and predicted it to have about 20% [alpha]-helix, 37% ß-strand, 11% turn, and 32% other by a four-component analysis. Prior analysis suggested at least 16% [alpha]-helix and 19% ß-sheet (7) in bovine IRBP. Thus, our expectations for the various proteins, whether insect cell-produced or E. coli-produced, if folded correctly, were values similar to those above.
The CD spectra are shown in Figure 4. For all the scans, a maximum ellipticity is found at 192 nm and a minimum at 208 nm. A local valley or inverted shoulder is found at 222 nm in all the spectra. The general shape of all the curves is basically the same, with most variation due to changes in magnitude that likely originate from differences in purity and protein concentration (the latter was roughly estimated based on A205). The spectra were deconvoluted according to several methods, all of which gave similar results as shown in Table II. These results are also similar to those mentioned above from bovine and recombinant human IRBP. Qualitatively, the CD spectra reveal no gross changes in conformation among the wild type and variants of IRBP. The CD spectra show similarities to spectra obtained for Tsp (18), which bears sequence homology with Domain B of IRBP repeats. These results suggest that the variant proteins and wild type all possess the same gross conformation, probably an [alpha]/ß or [alpha] + ß tertiary class protein (42, 43). In summary, the CD spectra show a common conformation of the repeats, even those produced in the two different expression systems. This validates their comparison in further experiments.
Fluorescence Excitation and Emission Wavelength Scans
The binding of ligands to IRBP frequently results in a several-fold enhancement of fluorescence (3, 7, 14, 30, 32). The excitation and emission characteristics of the IRBP-retinol and IRBP-16-AP complexes are shown in Figs. 5 and 6, respectively. The excitation and emission maxima for retinol-IRBP are at 331 and 479 nm (Figure 5). Similarly, the maxima for 16-AP-IRBP are 363 and 432 nm (Figure 6), respectively. There are no major differences among the wavelength scans for each ligand, except variations in amplitude, probably due to differing amounts of binding sites in the cuvettes. There is one notable change in the relative peak heights among the variants and the wild type protein in the emission scans at 430 nm for retinol (Figure 5), where a shoulder on the scans is slightly more prominent than in wild type. Other than this slight change, the identical wavelengths of the peaks suggest that the bound ligand is in a very similar hydrophobic environment irrespective of the proteins, variants or wild type. The variation in amplitude could be from a variation in protein concentration obtained by A280 measurements (28), though within experimental error (coefficient of variation [CV] ~5%) we adjusted it to 1 µM. Another explanation of the variation in peak height is the number of binding sites per polypeptides. Third, the inherent quantum yield of the ligand bound to each distinct binding site is simply different. To summarize, the spectra in Figs. 5 and 6 all show fluorescence enhancement, implying that the ligands bind to all these proteins and move less when bound.
Binding Properties: Measurements of Binding Capacities and Dissociation Constants
Within the context of this paper we define specific binding as a binding interaction that is saturable and possesses a high affinity. The raw fluorescence enhancement titration data are shown in Figure 7 for all-trans-retinol and in Figure 8 for 16-AP. Examination of the raw data shows that the wild type protein-ligand complex fluoresces more than the others, and the data show relatively little scatter. Nonlinear regression analysis of the binding curves with five parameters (Fo, S, e, Kd, and N) defined in the methods section yields estimates shown in Table IIIa for retinol and Table IIIb for 16-AP. The tables also show the value of Bmax per unit concentration of protein, which was calculated as the product of N and S. In some instances, it was necessary to compensate for the purity of the protein preparation of insect-produced protein. While the dissociation constants are all low, differences are found among S, N, and Bmaxs. The median coefficients of variation (CVs) of the measurements are shown in the bottom row of each table. These CVs suggest that the curve fitting approach works well. The CVs found for Fo, e, and Kd were somewhat high, as expected, since these measurements are very small.
The number of equivalent retinol binding sites increases with the length of the polypeptide in the insect cell expression series, from about 0.6 binding sites in R1 and R12- incrementally to about 1.3 binding sites in wild type protein. Thus, increasing the length of the polypeptide at the C-terminal end increases binding capability, thus supporting the hypothesis that each repeat can bind a molecule of retinol. However, this hypothesis suggests that as the length increases, the binding sites should increase from one site in R1 to two sites in R12+, three sites in R123, and four sites in wild type. These values (0.6 to 1.3) are consistent with the idea that each repeat can bind retinol, but the non-integral numbers and the likelihood of differences in fluorescence enhancement slope (S) among the different repeats (see below) suggested the need to construct and analyze each repeat individually. We next expressed each repeat separately in E. coli and analyzed each protein for its individual retinol-binding characteristics. Each repeat possesses the capacity to bind retinol. The number of binding sites for the individual repeats ranges from about 0.7 to 1.3 as shown in Table IIIa. Clearly, the data support our central hypothesis that each separate repeat contains a binding site.
The S-values of the individual repeats vary 4-fold for retinol and 20-fold for 16-AP. This suggests that the quantum yields of one ligand when bound to individual repeats are different. However, the trend is that Repeats 1 and 2 have the high S-values, while Repeats 3 and 4 are both lower. The variations in S among the repeats probably reflect differences in damping of motion of the ligand in the binding site, differences in the size and shape of the binding sites, differences in amino acid side chains in or near the binding sites, but not so much changes in hydrophobicity among the binding sites. Trivial explanations are also possible, for example, errors in protein concentration (though CVs are ~5%) or "bad" or inactive protein preparations.
The sum of the binding capacities of the four individual repeats is a little greater than binding capacity of WT for retinol. The summation of the Bmaxs from EcR1 through EcR4 is 16,300 ± 1170 SD versus the Bmax of wild type (10,200 ± 3330 SD). This difference is significant at p<0.01. Explanations of this observation are presented in the Discussion.
The above analyses have focused on individual repeats and nested deletions of IRBP. We also constructed two point mutations, which were suggested to us by McGee and Dryja (44) who found these sequence variations in patients, though it is probable that each change is not etiologic. We became interested in these variants because of the location, conservation, and chemical nature of the amino acid substitution. The point mutation, R725C, behaves very much like the wild type protein in number of binding sites, Kd, and Bmax per unit concentration of protein. On the other hand, G719S has a significantly smaller value for N and Bmax. These changes suggest that position 719 is very near a binding site or that glycine at this position is required for proper protein folding, whereas, position 725, though not far away, may not possess the same retinol binding interactions or the same structural attributes.
To summarize the retinol binding data, we find retinol fluorescence enhancement for each of the proteins that we constructed. For each protein there was a saturable high affinity interaction, implying one or more specific binding sites. The pattern of binding, N, Kd, and Bmaxs are all consistent with the central hypothesis of each repeat containing at least one binding site.
Fatty Acid Analog Binding Data
In comparing our WT binding data to Putalina et al (45), their N (1.1) from bovine IRBP is the same as ours, although they found a higher Kd of 0.36 µM. Also, 16-AP binds well to the human variants as shown in Table IIIb. EcR3 gives the lowest values for S, N, and Bmax, with EcR4 values a little higher. EcR2 values are the highest with EcR1 a little lower.
The 16-AP binding data argue against the notion that cryptic binding sites are exposed in the individually expressed repeats. The sum of the Bmaxs (23800 ± 8350 SD) from the four individual repeats is the same as the Bmax of the intact wild type molecule (23400 ± 9500 SD). The 16-AP binding data show proportionate increases in N, from 0.25 to 1.1, as the length of the polypeptide increases from about 300 to 1200 amino acids in the nested truncation series. This is consistent with a model of fatty acid binding in which each repeat independently binds 16-AP.
Regarding the fractional values of N found for the individual repeats, we suggest several possible explanations: 1. The binding sites may already be partially occupied by another ligand or molecule that may have contaminated the protein preparation: If there is a lot of fatty acid such as palmitate already bound to IRBP, then we fully expect that 16-AP would yield a fractional value of N. 2. Fractional N values may reflect a binding site that flickers between a binding and nonbinding state. This could occur if a physiologically relevant allosteric regulator changes the binding site conformation. 3. Some of the protein may be in a native state and functional whereas fractions of the protein may be inactive, damaged, or denatured. 4. Nonideal assay conditions may not mimic normal eye physiology. The pH, temperature, ionic conditions, osmotic strength, etc might profoundly affect N. 5. Less than 100% pure ligands may appear to reduce N.
To summarize, as shown in Figure 9, the fluorescence enhancement data suggest that each repeat can bind retinol and 16-AP without the need for additional polypeptide sequences from other repeats. Second, the point mutation data suggest that selected amino acids can profoundly alter binding capacity, apparently all in the absence of gross conformational changes. Third, models of retinol binding should consider the impact of the known gross shape changes in WT protein plus or minus retinol (46).
Discussion
IRBP as a Member of an Enzyme Family
A clear statistical argument can be made that the multiple sequence alignment and the resulting most parsimonious dendritogram represent divergence from a single common ancestor (20). The family includes members from all biological kingdoms. While the similarity is weak, we believe that it is legitimate to call these homologous relationships based on common ancestry. All these proteins bind ligands or catalyze substrates that are small and hydrophobic in character, suggesting some shared functional characteristics as well as structural and ancestral. In Tsp, Domain B possesses catalytic activity. Excepting IRBP, each family member contains only one Domain B per polypeptide; consequently, we predicted that each IRBP repeat can function as a monomeric binding protein, independent of the need for additional domains from other repeats. Our data support this prediction.
IRBP Production in Insect Cells
IRBP is synthesized (47, 48) and secreted from the photoreceptor cell. Wild type recombinant human IRBP is glycosylated by insect cells, secreted from them, and the engineered protein accumulates in the extracellular medium much as IRBP does in the IPS. The protein is co-translationally processed to remove the signal peptide in insect cells, and altered IRBP proteins can be evaluated initially by whether they are secreted, glycosylated, and N-terminally processed from insect cells (30).
Six of the seven baculovirus-produced proteins had the N-terminal signal peptide removed. In the seventh, R1, the protein retains its signal peptide. Potential explanations of the retention of the signal peptide by R1 include: 1. a sequence change in the protein, 2. the protein enters the medium after cell lysis, 3. the amount of secreted protein overwhelms the insect cell signal peptidase leaving the majority with the signal peptide intact. Countering the first point, we sequenced the recombinant baculovirus DNA and verified that no mutations had occurred in the R1 DNA sequence. Regarding the second point, we observe no obvious cell death in the cultures by microscopy, suggesting that this point cannot explain the observations. Possibility #3 seems the most likely alternative.
R123 and R12- are retained inside the insect cell despite cotranslational signal peptide removal and glycosylation. Possibly the C-terminal part of wild type IRBP is involved in subcellular trafficking. The lack of secretion may be analogous to opsin mutant proteins in RP retained in the rER or Golgi apparatus (70). Another explanation is that the posttranslational machinery of the insect cells is overwhelmed by the amounts of R123 and R12-. In spite of these deviations from the expected secretory pattern, these proteins have folded properly, as shown by CD analysis, and are useful in characterizing IRBP binding properties.
Immunological Properties
Donoso and group clearly showed that the monoclonal antibody, H3B5, reacts exclusively with the sequence AASEDPR, positions 356-362 found in Repeat 2 as an oligopeptide in a competition assay (27, 49). We were surprised to find that R1, which contains a related sequence SSLNDPR (positions 58 to 64), also cross-reacts with the antibody. We propose that the protein may have partially renatured while bound to the nitrocellulose blot. This raises the possibility that secondary and tertiary structure play a role in the binding of H3B5 to an epitope of the expressed protein. Our polyclonal antibody, GALS, shows cross-reaction among repeats 1, 3, and 4 suggesting a similar shape of epitopes in these three repeats. These immunologic cross-reactivities highlight the inherent similarity of structure among Repeats 1 through 4.
The Conformation of IRBP and Variants
CD analysis suggests that the use of E. coli and insect cells to express IRBP protein fragments is acceptable and results in native protein. The spectra of insect-produced R1 and E. coli-produced R1 are very similar. Also, the refolded EcR2 protein has a CD spectrum very similar to the wild type spectrum, showing that a denatured single repeat can direct its own refolding in vitro. All the proteins, expressed in insect cells or E. coli, fold in a grossly normal manner, and all appear to have the same general conformation. The results here are also very similar to the CD spectrum of bovine IRBP (7, 37). The CD spectra suggest that much of the protein folds into [alpha]-helices and ß-strand structures, and the general tertiary class may be [alpha] + ß as predicted by def_class.exe (42). This result suggests that IRBP is very different in conformation from other retinoid binding proteins, which are beta clamshell proteins. The only other protein related to IRBP, for which a CD analysis has been done, is Tsp, and its CD spectrum also suggests that it is an [alpha] + ß protein: The 208 nm valley is somewhat deeper than the 221 nm valley or inverted shoulder (18) in the CD spectrum. Manavalan and Johnson (43) suggest that [alpha] + ß proteins exhibit this behavior, whereas the [alpha]/ß tertiary class exhibit a deeper valley at 221 than at 208 nm. Deconvolution of CD spectra into various secondary structure components was carried out with several programs, each of which has its merits (50). An average structure of the 11 different proteins was obtained. Although unremarkable, the 18% [alpha]-helix, 30% ß-strand, 15% turn and 33% other (the remaining 4% is unpredicted), is most consistent with an [alpha] + ß or possibly [alpha]/ß tertiary class. While a caveat could be offered that a protein produced in different biological organisms as diverse as E. coli, insect cells, and humans might not possess the same conformation, these CD analyses suggest that it is not the case here.
Ligand Binding Properties
Our previous studies concluded that the ligand binding properties of human IRBP are similar to those of bovine IRBP (30). Also, fatty acid analogs bind even more tightly than retinoids to IRBP (51). These experiments suggested different roles for IRBP in transport ligands, and that there may be different structures in IRBP that bind these different ligands.
In this report, we only considered the sites involved in fluorescence enhancement. We chose the enhancement assay because of its rapidity, ease, reliability, and long history in IRBP work. Other assays share many of these same attributes, but most of these have additional complications. For example, the 3H-retinol binding assay is expensive, and we must then quantitatively separate free 3H-retinol from bound. While this can be achieved, free and bound retinol can stick to the DEAE column or charcoal (irreversibly), and the bound retinol can dissociate from IRBP during separation (58). Tryptophan fluorescence quenching is very useful, but detects two classes of binding sites in IRBP. For the purposes of this paper, we opted to examine the simpler, single-binding class behavior detected by the fluorescence enhancement assay.
Using this assay, the retinol dissociation constants from the various polypeptides all appear to have lower Kds than wild type, except EcR1, which is the same. The fluorescence enhancement slope (S) from EcR1 is the highest of the repeats suggesting that the fluorescence signal from this repeat dominates the response of the whole protein, and the effects from the other three repeats, because of their similar Kds and lower S-values probably appear as just a minor binding component in the analysis of the wild type protein's binding properties.
There is a significant difference between the Kd for retinol bound to WT (Kd = 1.04) in our earlier publication (30) and Kd reported here (Kd = 0.356, p <.05). The discrepancy probably occurs because: 1. The earlier value was derived from a limited data set, n=3. The new value is based on n=18. 2. Error is more definitively calculated here because of the numerical method (32). 3. The old method assumed ideal 1:1 binding behavior and a perfect hyperbola in plotting free versus bound. 4. The old method did not consider the inner filter effect or photobleaching of the ligand in the assay in the previous measurements. 5. There are some other variations such as different fluorometers and pipettors were used, and the cuvettes were at slightly different temperatures.
Ligand Binding with 16-AP
Studies (Table IIIb) with the binding of 16-AP suggest that each repeat binds this ligand. All the deletion proteins retained the ability to bind at least some retinol and 16-AP, suggesting that not all of the protein is needed to carry out the proposed essential functions of IRBP. The results of the point substitution, R725C, suggest that this change has minimal impact on ligand binding. However, the other point mutation, G719S, suggests that this change reduces retinol but not 16-AP binding. This suggests that this mutation is located in a retinol ligand binding site, but the binding of 16-AP does not require glycine at position 719.
Previous studies have characterized fatty acid binding to bovine IRBP. Bazan et al (52) found up to four fatty acids noncovalently attached and two covalently attached. Putalina et al. (45) used 16-AP and other fluorescent fatty acids and found one independent binding site. Our data here, based on individual human repeats, suggest that there are four distinct 16-AP sites, all sharing similar ligand binding properties.
The fatty acid binding sites may have essential roles in the function of IRBP in the Visual Cycle. Chen et al (53-55) propose that an IRBP-11-cis-retinal complex selectively dissociates when docosahexaenoic acid (DHA) binds to the protein near the photoreceptor cell. DHA has been shown to be released from rat retina in response to light (56). Although there are some qualifications, such as the DHA release was measured from whole retina and was not a measurement of DHA released solely into the IPS, and DHA release was not simultaneously measured from the RPE, these experiments still lend support to the idea (53) that DHA is a regulator of retinoid transport. Where could the DHA binding site be in comparison to the retinol binding sites? One possibility is that one repeat possesses a DHA binding site, while another repeat binds all-trans-retinol. Another possibility is that Domain A in each repeat may contain an allosteric affector site whereas the binding site for retinoids or other fatty acids is in Domain B. Future experiments will measure protein fluorescence of Domains A and B and mutants thereof to examine whether a regulatory class of binding sites is located in Domain A and a different (more hydrophobic) binding site resides in Domain B.
Some differences in the Bmax of WT and the individual repeats might be attributed to different fatty acids, in particular DHA, bound to the various proteins. The WT IRBP was produced in insect cells. The media for these cells do not contain DHA or linolenic acid, although traces could be derived from the limited amount of fetal calf serum present during expression. Thus, even though we did not strip the proteins by lipidex treatment, it is not likely that there is much DHA bound to insect-produced WT IRBP. E. coli does not produce DHA, and the medium does not contain any either. The DHA competitive effects on retinol fluorescence enhancement of Chen et al. (53) were small (less than 18%) and required about a five-fold molar excess of DHA over protein. [Other fatty acids showed smaller effects (54)]. Given that these levels of DHA are 10 times the normal physiological level of DHA bound endogenously to IRBP, and given that there probably is far less DHA bound to our WT IRBP preparations, potential differences in endogenous levels of DHA between the insect and E. coli expressed proteins should not markedly change the interpretation of our results.
Linearization versus Numerical Methods of Binding Curve Analysis
Disagreement in measuring N, Bmax, and Kd among various reports of fluorescence enhancement of retinol bound to IRBP may in part be due to the numerical analysis methods employed. The Cogan plot (57) was designed to provide a linear plot, which is ideal for visualization of the data. This makes it easy to eyeball a suitable line or to use a least squares linear regression analysis of the data, which are simple to calculate. However, there are several potential problems with this analysis, which are more fully discussed elsewhere (32, 58, 59). To summarize the problems: 1. Eyeballing a line is subjective. 2. The Cogan plot frequently yields data that look a little sigmoidal: Can we justify approximating this sigmoidal data with a straight line? What criteria could we use to decide on a valid range of ligand concentrations to avoid the nonlinear tails? 3. Data are obtained as total retinol concentration (the independent variable) versus fluorescence (the dependent variable). The Cogan analysis plots two quantities against each other, neither of which is independent of the other (Po[alpha] vs. Ro([alpha]/(1-[alpha]))), and all X- and Y-values are affected by estimates of Fo and Fmax. This can cause complications in estimates of error (60). 4. The Cogan plot derives Kd and N from the slope of the line and extrapolation to the Y-intercept. Extrapolations can magnify error. In contrast, the Baer method (32) relies on a nonlinear regression numerical method. It avoids all of the above-mentioned problems. Despite all these issues, Chen et al. (53) found only a 20% difference in their values for Kd in comparing the Cogan analysis versus a numerical approach (58).
Baer et al. (32) introduced this elegant way of treating fluorescence enhancement titration data, but they did not measure the number of binding sites in full-size IRBP. Thus, satisfactory answers about the number of binding sites have awaited methods to examine all the structural elements in IRBP. Repeat 4 in Xenopus contains a binding site of all-trans-retinol with N = 0.15 (32) (though they presented higher values recently (61)). Previously, we showed retinol-binding in a variant protein composed of Repeats 1, 2, and part of 3 (62). These results suggested that there are at least two structurally distinct retinol binding sites in IRBP. Also, some fish IRBP proteins contain only two repeats (63, 64), though we don't know for certain whether these fish IRBPs bind retinoids. All these results suggest multiple ligand binding sites in intact IRBP, and none exclude the possibility of four retinol or fatty acid binding sites in the whole protein.
Utility of Expression Approaches for Molecular Etiology
It is impossible to obtain mutated IRBPs from human eyes directly, and IRBPs purified from other species are imperfect substitutes: They may contain so many compensatory amino acid changes that it would confound analysis even if we found a species with a putative gene lesion. But, via expression systems and with human cDNA clones, we have begun a molecular dissection of the human IRBP protein. The truncated proteins that we made retain many structural and functional characteristics of IRBP but with reduced binding capacities. The proteins also are stable in the insect cells with many fully secreted. Thus, they do not cause gain-of-function toxicity in these cells. However, some proteins are secreted while others are not. The sequence change of G719S (in Domain B of Repeat 3) found by McGee et al. (44) may be a subclinical point mutation, as it appears to reduce N by 29% (0.923 vs. 1.30) when compared to WT protein. Given the identified reduced binding capacities of the variants and the secretion problems described here, IRBP should now be considered as a candidate for inherited retinal diseases.
Comparison of R1 and EcR1
Why should there be a disparity between the ligand binding properties of R1 and EcR1? While the slope factor (S) and the extinction coefficient (e) for each of these two very closely related proteins are the same, the number of binding sites (N) and the Kds are different. There are several potential causes of the differences: 1. The signal peptide is retained in R1 whereas the N-terminal sequence of EcR1 is different and contains seven vector-derived amino acids. These sequences are most likely to interact with Domain A, a candidate regulatory domain. 2. R1 has attached carbohydrate, while EcR1 has none. 3. The C-terminal ends are slightly different. 4. Differences in the quality of the preparations. 5. Perhaps not all of the R1 protein is folded normally. 6. The protein concentration of R1 may be lower than that of EcR1. Jointly these points may form a plausible explanation of the differing ligand binding properties.
Negative Cooperativity?
The "lack of additivity" between the sum of Bmax from the four individual repeats and WT IRBP causes us to speculate that negative cooperativity (65) plays a role in the transport of retinoids across the IPS. Negative cooperativity would suggest that one ligand bound to IRBP may directly or indirectly affect the binding of additional ligands. This may imply that the binding sites in IRBP may change conformation or come into close proximity by bending of the protein once the first ligand molecule is bound. Adler et al. (46) noticed a distinctive shape change in IRBP upon retinol binding.
Another possible explanation of the "lack of additivity" is that the first binding event makes a conformational shape change in WT that reduces the fluorescence enhancement properties of the other three binding sites, without altering their affinities for retinol. There are other potential explanations but these appear less plausible: 1. Cryptic nonfunctional binding sites are exposed and activated once the protein is subdivided into individual repeats, but the 16-AP binding data argue against this. 2. The wild type protein preparations might not be as "good" as the E. coli-derived preparations. Countering the latter argument similar numbers of hydrophobic retinol binding sites per polypeptide (N ranging from 0.55 to about 1) have been reported for intact bovine IRBP by absorbance (7) and fluorescence enhancement (12, 54).
The putative negative cooperativity in IRBP function could be analogous to CO2 causing the sudden release of four O2 molecules from hemoglobin: After a light bleach all-trans-retinol or DHA may be present at high concentration near the photoreceptor plasma membrane. These may allosterically affect IRBP, causing the release of 11-cis-retinal near the same membrane, but only when a demand for the aldehyde presents itself. Thus, IRBP, may serve as a reservoir for 11-cis-retinal that can be tapped as required.
Conclusions
Small IRBP fragments fold into a normal IRBP conformation and can bind substantial amounts of retinoid and fatty acid analogs. Binding curve analyses show these interactions to be high in affinity and saturable, implying that these are specific binding phenomena. Our data rule out models of IRBP action that require the whole or even half the protein to bind one ligand. Each individual repeat binds retinol and 16-AP, suggesting that a monomeric repeat unit can bind different ligands and a single repeat can be multifunctional in its binding capabilities. Similar results were found with each repeat (none was functionally dead), though some variation in quantum efficiency may explain apparent differences in S, N, and Bmax.
Acknowledgements
These studies were supported in part by the Fight for Sight Research Division of the National Society to Prevent Blindness (grant-in-aid GA92017); Research to Prevent Blindness, Olga Keith Weiss Award; and the National Eye Institute, P30 EY6360, R01 EY 10553.
References
1. Bunt-Milam AH, Saari JC. Immunocytochemical localization of two
retinoid-binding proteins in vertebrate retina. J Cell Bio 1983;
97:703-712. ![]()
2. Carlson A, Bok D. Promotion of the release of 11-cis-retinal from
cultured retinal pigment epithelium by interphotoreceptor
retinoid-binding protein. Biochemistry 1992; 31:9056-9062.
![]()
3. Okajima TI, Pepperberg DR, Ripps H, Wiggert B, Chader GJ.
Interphotoreceptor retinoid-binding protein: role in delivery of retinol
to the pigment epithelium. Exp Eye Res 1989; 49:629-644.
![]()
4. Crouch RK, Hazard ES, Lind T, Wiggert B, Chader G, Corson DW.
Interphotoreceptor retinoid binding protein and alpha-tocopherol
preserve the isomeric and oxidation state of retinol. Photochem
Photobiol 1992; 56:251-255. ![]()
5. Adler AJ, Edwards RB. IRBP uniquely removes 11-cis-retinal from isolated RPE membranes. Invest Ophthalmol Vis Sci 1995; 36:S6.
6. Ho MT, Massey JB, Pownall HJ, Anderson RE, Hollyfield JG.
Mechanism of vitamin A movement between rod outer segments,
interphotoreceptor retinoid-binding protein, and liposomes. J Biol Chem
1989; 264:928-935. ![]()
7. Adler AJ, Evans CD, Stafford WF 3d. Molecular properties of bovine
interphotoreceptor retinol-binding protein. J Biol Chem 1985;
260:4850-4855. ![]()
8. Saari JC, Bunt-Milam AH, Bredberg DL, Garwin GG. Properties and
immunocytochemical localization of three retinoid-binding proteins from
bovine retina. Vision Res 1984; 24:1595-1603. ![]()
9. Beuckmann CT, Gordon WC, Kanaoka Y, Eguchi N, Marcheselli VL,
Gerashchenko DY, Urade Y, Hayaishi O, Bazan NG. Lipocalin-type
prostaglandin D synthase (beta-trace) is located in pigment epithelial
cells of rat retina and accumulates within interphotoreceptor matrix. J
Neurosci 1996; 16:6119-6124. ![]()
10. Tanaka T, Urade Y, Kimura H, Eguchi N, Nishikawa A, Hayaishi O.
Lipocalin-type prostaglandin D synthase (beta-trace) is a newly
recognized type of retinoid transporter. J Biol Chem 1997;
272:15789-15795. ![]()
11. Pfeffer B, Wiggert B, Lee L, Zonnenberg B, Newsome D, Chader G.
The presence of a soluble interphotoreceptor retinol-binding protein
(IRBP) in the retinal interphotoreceptor space. J Cell Physiol 1983;
117:333-341. ![]()
12. Okajima TI, Pepperberg DR, Ripps H, Wiggert B, Chader GJ.
Interphotoreceptor retinoid-binding protein promotes rhodopsin
regeneration in toad photoreceptors. Proc Natl Acad Sci U S A 1990;
87:6907-6911. ![]()
13. Jones GJ, Crouch RK, Wiggert B, Cornwall MC, Chader GJ. Retinoid
requirements for recovery of sensitivity after visual-pigment bleaching
in isolated photoreceptors. Proc Natl Acad Sci U S A 1989; 86:9606-9610.
![]()
14. Pepperberg DR, Okajima TL, Wiggert B, Ripps H, Crouch RK, Chader
GJ. Interphotoreceptor retinoid-binding protein (IRBP). Molecular
biology and physiological role in the visual cycle of rhodopsin. Mol
Neurobiol 1993; 7:61-85. ![]()
15. Stanhope MJ, Czelusniak J, Si JS, Nickerson J, Goodman M. A
molecular perspective on mammalian evolution from the gene encoding
interphotoreceptor retinoid binding protein, with convincing evidence
for bat monophyly.. Mol Phylogenet Evol 1992; 1:148-160.
![]()
16. Stanhope MJ, Bailey WJ, Czelusniak J, Goodman M, Nickerson J, Si JS, Singer GA, Kleinschmidt TK. A molecular view of primate supraordinal relationships from the analysis of both nucleotide and amino acid sequences. In: MacPhee RD, ed., Primates and Their Relatives in Phylogenetic Perspective, New York: Plenum Press; 1993:251-292.
17. Shestakov SV, Anbudurai PR, Stanbekova GE, Gadzhiev A, Lind LK,
Pakrasi HB. Molecular cloning and characterization of the ctpA gene
encoding a carboxyl-terminal processing protease. Analysis of a
spontaneous photosystem II-deficient mutant strain of the cyanobacterium
Synechocystis sp. PCC 6803. J Biol Chem 1994; 269:19354-19359.
![]()
18. Keiler KC, Sauer RT. Identification of active site residues of
the Tsp protease. J Biol Chem 1995; 270:28864-28868. ![]()
19. Silber KR, Keiler KC, Sauer RT. Tsp: A tail-specific protease
that selectively degrades proteins with nonpolar C termini. Proc Natl
Acad Sci U S A 1992; 89:295-299. ![]()
20. Nickerson JM, Goodman M, Lin ZY, Czelusniak J, Mian IS. Predicted structure, function, and evolution of the repeated domain present in interphotoreceptor retinoid-binding protein (IRBP) and its homologs. Invest Ophthalmol Vis Sci 1997; 38:S3.
21. Borst DE, Nickerson JM. The isolation of a gene encoding
interphotoreceptor retinoid-binding protein. Exp Eye Res 1988;
47:825-838. ![]()
22. Borst DE, Redmond TM, Elser JE, Gonda MA, Wiggert B, Chader GJ,
Nickerson JM. Interphotoreceptor retinoid-binding protein. Gene
characterization, protein repeat structure, and its evolution. J Biol
Chem 1989; 264:1115-1123. ![]()
23. Si JS, Borst DE, Redmond TM, Nickerson JM. Cloning of cDNAs
encoding human interphotoreceptor retinoid-binding protein (IRBP) and
comparison with bovine IRBP sequences. Gene 1989; 80:99-108.
![]()
24. Lin ZY, Si JS, Phong M, Li GR, Nickerson JM. Human IRBP and its deletion mutants. Invest Ophthalmol Vis Sci 1996; 37:S908.
25. Lin ZY, Li GR, Phong M, Si JS, Takizawa N, Nickerson JM. Study of recombinant human IRBP and its mutations. Invest Ophthalmol Vis Sci 1997; 38:S3.
26. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Press; 1989:18.60-18.75.
27. Donoso LA, Merryman CF, Sery TW, Vrabec T, Arbizo V, Fong SL.
Human IRBP: characterization of uveitopathogenic sites. Curr Eye Res
1988; 7:1087-95. ![]()
28. Gill SC, von Hippel PH. Calculation of protein extinction
coefficients from amino acid sequence data. Anal Biochem 1989;
182:319-326. [Erratum Anal Biochem 1990; 189:283] ![]()
29. Vialard J, Lalumiere M, Vernet T, Briedis D, Alkhatib G, Henning
D, Levin D, Richardson C. Synthesis of the membrane fusion and
hemagglutinin proteins of measles virus, using a novel baculovirus
vector containing the beta-galactosidase gene. J Virol 1990; 64:37-50.
![]()
30. Lin ZY, Si JS, Nickerson JM. Biochemical and biophysical
properties of recombinant human interphotoreceptor retinoid binding
protein. Invest Ophthalmol Vis Sci 1994; 35:3599-3612. ![]()
31. Summers MD, Smith GE. A manual of methods for baculovirus vectors and insect cell culture procedures. College Station: Texas Agricultural Station; 1988:Bulletin # 1555.
32. Baer CA, Kittredge KL, Klinger AL, Briercheck DM, Braiman MS,
Gonzalez-Fernandez F. Expression and characterization of the fourth
repeat of Xenopus interphotoreceptor retinoid-binding protein in E.
coli. Curr Eye Res 1994; 13:391-400. ![]()
33. Jin Y, Cerletti N. Western blotting of transforming growth factor
beta 2. Optimization of the electrophoretic transfer. Appl Theor
Electrophor 1992; 3:85-90. ![]()
34. Matsudaira P. Sequence from picomole quantities of proteins
electroblotted onto polyvinylidene difluoride membranes. J Biol Chem
1987; 262:10035-10038. ![]()
35. Woody RW. Circular Dichroism. Methods Enzymol 1995; 246:34-71.
![]()
36. Johnson WC Jr. Protein secondary structure and circular
dichroism: a practical guide. Proteins 1990; 7:205-214.
![]()
37. Hazard ES, Lin ZY, Nickerson JM, Wiggert B, Chader G, Crouch RK. A circular Dichroism (CD) analysis of IRBP. Invest Ophthalmol Vis Sci 1994; 35:1464.
38. Provencher SW, Glockner J. Estimation of globular protein
secondary structure from circular dichroism. Biochemistry 1981;
20:33-37. ![]()
39. Sreerama N, Woody RW. Protein secondary structure from circular
dichroism spectroscopy. Combining variable selection principle and
cluster analysis with neural network, ridge regression and
self-consistent methods. J Mol Biol 1994; 242:497-507. ![]()
40. Sreerama N, Woody RW. A self-consistent method for the analysis
of protein secondary structure from circular dichroism. Anal Biochem
1993; 209:32-44. ![]()
41. Manavalan P, Johnson CW Jr. Variable selection method improves
the prediction of protein secondary structure from circular dichroism
spectra. Anal Biochem 1987; 167:76-85. ![]()
42. Venyaminov SY, Vassilenko KS. Determination of protein tertiary
structure class from circular dichroism spectra. Anal Biochem 1994;
222:176-184. ![]()
43. Manavalan P, Johnson CW. Sensitivity of circular dichroism to protein tertiary structure class. Nature 1983; 305:831-832.
44. McGee TL, DeStefano JD, Berson EL, Dryja TP. A complete screen of the entire coding sequence of the IRBP gene for mutations in patients with hereditary retinal degenerations. Invest Ophthalmol Vis Sci 1993; 34:1460.
45. Putilina T, Sittenfeld D, Chader GJ, Wiggert B. Study of a fatty
acid binding site of interphotoreceptor retinoid-binding protein using
fluorescent fatty acids. Biochemistry 1993; 32:3797-3803.
![]()
46. Adler AJ, Stafford WF 3d, Slayter HS. Size and shape of bovine
interphotoreceptor retinoid-binding protein by electron microscopy and
hydrodynamic analysis. J Biol Chem 1987; 262:13198-13203.
![]()
47. Porrello K, Bhat SP, Bok D. Detection of interphotoreceptor
retinoid binding protein (IRBP) mRNA in human and cone-dominant squirrel
retinas by in situ hybridization. J Histochem Cytochem 1991; 39:171-176.
![]()
48. van Veen T, Katial A, Shinohara T, Barrett DJ, Wiggert B, Chader
GJ, Nickerson JM. Retinal photoreceptor neurons and pinealocytes
accumulate mRNA for interphotoreceptor retinoid-binding protein (IRBP).
FEBS Lett 1986; 208:133-137. ![]()
49. Donoso LA, Rodrigues M, Vrabec TR, Sery TW, Fong SL. IRBP:
preparation and characterization of site-specific monoclonal antibodies.
Curr Eye Res 1990; 9:357-362. ![]()
50. Greenfield NJ. Methods to estimate the conformation of proteins
and polypeptides from circular dichroism data. Anal Biochem 1996;
235:1-10. ![]()
51. Nickerson JM, Mody V, DeGuzman C, Heron KA, Marciante K, Boatright J, Si JS, Lin ZY. Ligand-binding properties of recombinant human IRBP. In: Anderson RE, LaVail MM, Hollyfield JG, eds.,Degenerative Diseases of the Retina, New York: Plenum Press; 1995:395-402.
52. Bazan NG, Reddy TS, Redmond TM, Wiggert B, Chader GJ. Endogenous
fatty acids are covalently and noncovalently bound to interphotoreceptor
retinoid-binding protein in the monkey retina. J Biol Chem 1985;
260:13677-13680. ![]()
53. Chen Y, Houghton L, Brenna JT, Noy N. Docosahexaenoic acid
modulates the interactions of the interphotoreceptor retinoid-binding
protein with 11-cis-retinal. J Biol Chem 1996; 271:20507-20515.
![]()
54. Chen Y, Saari JC, Noy N. Interactions of all-trans-retinol and
long-chain fatty acids with interphotoreceptor retinoid-binding protein.
Biochemistry 1993; 32:11311-11318. ![]()
55. Chen Y, Noy N. Retinoid specificity of interphotoreceptor
retinoid-binding protein. Biochemistry 1994; 33:10658-10665.
![]()
56. Reinboth JJ, Clausen M, Reme CE. Light elicits the release of
docosahexaenoic acid from membrane phospholipids in the rat retina in
vitro. Exp Eye Res 1996; 63:277-284. ![]()
57. Cogan U, Kopelman M, Mokady S, Shinitzky M. Binding affinities of
retinol and related compounds to retinol binding proteins. Eur J Biol
1976; 65:71-78. ![]()
58. Norris AW, Cheng L, Giguere V, Rosenberger M, Li E. Measurement
of subnanomolar retinoic acid binding affinities for cellular retinoic
acid binding proteins by fluorometric titration. Biochim Biophys Acta
1994; 1209:10-18. ![]()
59. Birdsall B, King RW, Wheeler MR, Lewis CA Jr, Goode SR, Dunlap
RB, Roberts GC. Correction for light absorption in fluorescence studies
of protein-ligand interactions. Anal Biochem 1983; 132:353-361.
![]()
60. Johnson ML, Frasier SO. Nonlinear least-squares analysis. Meth Enzymol 1985; 117:301-342.
61. Baer CA, Van Neil EE, Braiman M, Gonzalez-Fernandez F. Truncational mutagenesis of the fourth module of Xenopus IRBP. Invest Ophthalmol Vis Sci 1997; 38:S3.
62. Redmond TM, Nickerson JM. Retinoid binding to a recombinant central two repeat segment of IRBP. Invest Ophthalmol Vis Sci 1992; 33:1181.
63. Rajendran RR, Van Niel EE, Stenkamp DL, Cunningham LL, Raymond
PA, Gonzalez-Fernandez F. Zebrafish interphotoreceptor retinoid-binding
protein: differential circadian expression among cone subtypes. J Exp
Biol 1996; 199:2775-2787. ![]()
64. Wagenhorst BB, Rajendran RR, Van Niel EE, Hessler RB, Bukelman A,
Gonzalez-Fernandez F. Goldfish cones secrete a two-repeat
interphotoreceptor retinoid-binding protein. J Mol Evol 1995;
41:646-656. ![]()
65. Voet D, Voet JG. Biochemistry. New York: John Wiley and Sons, Inc.; 1990:234-241.
66. Nickerson JM, Borst DE, Redmond TM, Si JS, Toffenetti J, Chader GJ. The molecular biology of IRBP: application to problems of uveitis protein chemistry, and evolution. In: Farber DB, Chader GJ, eds.,Molecular biology of the retina: Basic and clinically relevant studies, New York: Wiley-Liss, Inc.; 1991:139-161.
67. Harrison SC. Peptide-surface association: the case of PDZ and PTB
domains. Cell 1996; 86:341-343. ![]()
68. Redmond TM, Wiggert B, Robey FA, Chader GJ. Interspecies
conservation of structure of interphotoreceptor retinoid-binding
protein. Similarities and differences as adjudged by peptide mapping and
N-terminal sequencing. Biochem J 1986; 240:19-26. [Erratum Biochem J
1987; 242:935] ![]()
69. Fong SL, Cook RG, Alvarez RA, Liou GI, Landers RA, Bridges CD.
N-terminal sequence homologies in interstitial retinol-binding proteins
from 10 vertebrate species. FEBS Lett 1986; 205:309-12.
![]()
70. Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J.
Functional heterogeneity of mutant rhodopsins responsible for autosomal
dominant retinitis pigmentosa. Proc Natl Acad Sci U S A 1991;
88:8840-8844. ![]()