 Molecular Vision
2010; 16:1227-1240
<http://www.molvis.org/molvis/v16/a136>
Molecular Vision
2010; 16:1227-1240
<http://www.molvis.org/molvis/v16/a136> Received 5 April 2010 | Accepted 23 June 2010 | Published 6 July 2010
Molecular Vision
2010; 16:1227-1240
<http://www.molvis.org/molvis/v16/a136>
Received 5 April 2010 | Accepted 23 June 2010 | Published 6 July 2010
Naresh Polisetti,1 Prasoon Agarwal,2 Imran Khan,2 Paturu Kondaiah,2 Virender S. Sangwan,3 Geeta K. Vemuganti1
1Sudhakar and Sreekanth Ravi Stem Cell Biology Laboratory, C-TRACER, Hyderabad Eye Research Foundation, L.V. Prasad Eye Institute, Hyderabad, India; 2Molecular Reproduction, Development and Genetics, Indian Institute of Science, Banglore, India; 3Cornea and Anterior Segment Service, L.V. Prasad Eye Institute, Hyderabad, India
Correspondence to: Geeta K. Vemuganti, M.D., D.N.B., F.N.A.M.S., Head, Ophthalmic Pathology Service, Sudhakar and Sreekanth Ravi Stem Cell Biology Laboratory, Champalimaud Translational Centre for Eye Research, Brien Holden Eye Research Centre, Hyderabad Eye Research Foundation, L.V. Prasad Eye Institute, L.V. Prasad Marg, Banjara Hills, Hyderabad - 500 034, India; Phone: +91-40-30612505; FAX: +91-40-2354 8271; email: geeta@lvpei.org
Purpose: Limbal stem cell deficiency is a challenging clinical problem and the current treatment involves replenishing the depleted limbal stem cell (LSC) pool by either limbal tissue transplantation or use of cultivated limbal epithelial cells (LEC). Our experience of cultivating the LEC on denuded human amniotic membrane using a feeder cell free method, led to identification of mesenchymal cells of limbus (MC-L), which showed phenotypic resemblance to bone marrow derived mesenchymal stem cells (MSC-BM). To understand the transcriptional profile of these cells, microarray experiments were carried out.
Methods: RNA was isolated from cultured LEC, MC-L and MSC-BM and microarray experiments were carried out by using Agilent chip (4×44 k). The microarray data was validated by using Realtime and semiquntitative reverse transcription polymerase chain reaction.
Results: The microarray analysis revealed specific gene signature of LEC and MC-L, and also their complementary role related to cytokine and growth factor profile, thus supporting the nurturing roles of the MC-L. We have also observed similar and differential gene expression between MC-L and MSC-BM.
Conclusions: This study represents the first extensive gene expression analysis of limbal explant culture derived epithelial and mesenchymal cells and as such reveals new insight into the biology, ontogeny, and in vivo function of these cells.
One of the most important advances made in translational research is in the field of ocular surface reconstruction using cell therapy [1-3]. This technology owes its success not only to the surgical advances but also to the increasing amount of knowledge pertaining to the location, characteristics and functioning of Limbal stem cells (LSC) [4-6]. In the normal uninjured state, LSC are mitotically quiescent and maintained in a specialized limbal stromal microenvironment or “niche.” However, upon corneal epithelial wounding, stem cells located in the limbus proliferate to generate more stem cells and transient amplifying cells to replace the damaged epithelium. It is generally agreed that the LSC are characterized by special location in the limbus, clonality, cytokeratin profile, transformation-related protein 63 (p63) delta isomers, and ATP-binding cassette sub-family G member 2 (ABCG2) expression [7-9]. It is well established that the niche plays an important role in the maintenance of stem cell properties in several tissues and this is expected to be true in the case of the LSC niche as well [10-13]. Some of the assumed factors for niche regulation include proximity to vasculature [14]; the basement membrane composition with respect to specific isoforms of collagen IV, laminin and fibronectin [15]; and the presence of limbal fibroblasts in the underlying stroma, which produce various cytokines [16].
We had earlier reported the presence of spindle shaped cells in extended limbal explant cultures, which bear a striking resemblance to the mesenhcymal stem cells derived from bone marrow (MSC-BM), which we had referred to as mesenhcymal like cells from limbus (MC-L) [17]. Interestingly, limbal fibroblast-like cells have also been reported to have stem cell like properties [18] and their conditioned media has been reported to foster conversion of human embryonic stem cells into corneal epithelial-like cells [19].
Several groups have reported the gene expression profile of limbal and corneal epithelial cells that has significantly contributed to the understanding of several cellular pathways and intrinsic factors that underpin the phenotypic difference between the two cell types [20-22]. These studies and the study by Zhou et al. [23], have used the native corneal and limbal tissue to derive the gene expression profile. However the gene expression profile of the cultured human limbal epithelial and stromal cells cultured cells obtained from the native limbal tissue that is used for clinical transplantation to regenerate the ocular surface has not been addressed until now. In the present study, we evaluated the transcriptome of the limbal explant culture derived epithelial and mesenchymal like cells by microarray and identified expression of unique genes and biologic pathways that characterize both these cell types. To evaluate our hypothesis that the MC-L possibly act as one of the “niche” derived intrinsic feeder cells in the feeder cell free method of limbal explants culture, we compared the profile of these cells to that of the MSC-BM, which form the supporting niche for the hematopoietic system.
All the procedures, recruitment of patients and the protocol were approved by the Institutional Review Board (L.V. Prasad Eye Institute IRB, Hyderabad, India) and the research followed the tenets of the declaration of Helsinki.
In an ongoing clinical trial, which was approved by the IRB, limbal epithelial cultures were established from limbal biopsies as described in our previous publications [2,3,17]. Briefly, less than 1×2 mm2 piece of limbal biopsy was obtained from the limbal region which included the epithelium as well as 0.5 mm of stromal tissue. Limbal epithelial cultures were established on de-epitheliazed human amniotic membrane (dhAM) with the basement membrane side up, in a feeder cell-free culture system. Cultures were established in duplicates for each clinical sample of which one culture was used for transplantation (after 10–14 days in culture) while the other culture used for experiments. A total of 25 samples were used in this study of which, 3 samples were used for RNA isolation and the remaining were cultured further. The limbal explants cultures in the duplicate plates were incubated for 2–3 weeks more to propogate the adherent spindle cells in the bottom of the Petri dish, beyond the area of amniotic membrane, which contained epithelial cells (n=22). These plates adherent cells (epithelial and spindle cells) were then trypsinized and plated on a T25 flask (Thermofisher Scientific, Roskilde, Denmark) in DMEM medium supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich Inc., St. Louis, MO). The non-adherent epithelial cells suspended in the medium were removed by changing the medium. These cultures were maintained at 37 °C and 5% CO2 in humidified incubator (BINDER GmbH, Tuttlingen, Germany). When the cells reached 80%–90% confluence, cultures were harvested with 0.25% trypsin (Sigma-Aldrich) in 1mM EDTA solution (Sigma-Aldrich). MSC-BM cells were obtained using a previously described protocol [17]. Briefly, human MSC-BM cultures were established from bone marrow aspirates of healthy donors after obtaining informed consent. The bone marrow mononuclear cells (BMMNCs) were separated using Ficoll-Hypaque gradient at 400× g for 30 min. The mononuclear cells were then plated at a density of 1×107 cells in Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich Chemie, Steinheim, Germany) supplemented with 10% FBS. When cultures reached confluence, cells were passaged using trypsin-EDTA in line 100.
Immunocytochemical analysis-- Immunocytochemical analysis was performed as previously described [17]. Briefly, cells were incubated with primary antibodies for vimentin, ATP-binding cassette subfamily G member 2 (ABCG2), Cytokeratin 14 (KRT14), Cytokeratin 3/12 (KRT3/12), E-cadherin (CDH1), cytokeratin 19 (KRT19), cluster of differentiation 45 (CD45), and nestin (Chemicon, Billerica, MA). Double immunostaining was performed for vimentin and paired box gene 6 (PAX-6). Following incubation with secondary antibodies (conjugated to Fluorscein Isothiocynate (FITC) or Tetramethyl Rhodamine Isothiocynate (TRITC), the nucleus was counterstained with propidium iodide (PI). The stained preparations were screened with a laser scanning confocal microscope (LSM 510; Carl Zeiss, Jena, Germany) using a fluorescent light source.
Flow cytometry analysis-- MC-L were characterized for the expression of CD44-PE (Phycoerythrin), CD90-FITC, CD13-FITC, human leukocyte antigen (HLA-ABC-PE and HLA-DR-PE), CD10-PE, CD40-FITC, CD11b-FITC, CD40L-APC (Allophycocyanin), CXC chemokine receptor 4 (CXCR4) –APC (eBioscienceTM, San Diego, CA) CD34-FITC, CD138-PerCP (Peridinin Chlorophyll Protein Complex; BD Pharmingen™, San Diego, CA) markers. Briefly, a single cell suspension of 0.5 to 1×106 cells obtained in 100 µl of PBS (phosphate buffered saline) containing 0.1% sodium azide and 2% FBS. These were incubated with saturating concentrations of the respective primary antibodies or conjugated antibodies for 45 min. After three washes, the cells were centrifuged at 200× g for 5 min and resuspended in ice-cold PBS. Fluorescence was evaluated by BD-FACS Aria (BD Biosciences) and data were analyzed by using FACS Diva software. Corresponding isotype controls were included in each experiment and specific staining was measured from the cross point of the isotype with a specific antibody graph. A total of 20,000 events were acquired to determine the positivity of different cell surface markers used.
Total RNA was extracted from each sample (n=3 for LEC, MC-L, and MSC-BM) with Trizol (Invitrogen, Carlsbad, CA) and purified using RNAeasy Columns (Qiagen GmbH, Hilden, Germany,). RNA was quantitated by NanoDrop ND-1000 (Thermo Scientific, Wilmington DE) and the quantity and integrity was established by resolving on 0.8% formaldehyde agarose gels.
Microarray experiments were performed using Agilent 4×44k oligonucleotide arrays. These arrays cover the entire genome. For labeling reaction, 500 ng of RNA from LEC, MC-L, and MSC-BM was used (n=2 of each LEC, MC-L, and MSC-BM). Labeling was done using the Quick Amp labeling kit (Agilent technologies, Santa Clara, CA) as per the manufacturer’s protocol. Briefly, using T7 promoter element coupled oligodT primer, cDNA was generated and from cDNA, labeled cRNA was generated via an in vitro transcription reaction using T7 RNA polymerase and Cy3 (for LEC and MC-L) or Cy5 (MC-L and MSC-BM) CTP. Labeled cRNA (825 ng) of the respective sample was used for hybridization in the following combinations - LEC (Cy3) and MC-L (Cy5), LEC (Cy3) and MSC-BM (Cy5) and MC-L (Cy3) and MSC-BM (Cy5). Hybridization was performed for 17 h, rotating at a speed of 10 rpm at 65 °C in an hybridization oven (Agilent Technologies).
Microarray image analysis was done using Feature extraction version 9.5.3.1 (Agilent Technologies) and data analysis was done using Gene Spring version 10 (Agilent Technologies). The background corrected intensity values were used for analysis. Normalization was done using LOWESS algorithm. Similarly expressed genes were filtered on the basis of standard deviation between two biologic replicates with the cut off of less than one. Fold changes were calculated and genes with more than twofold difference were selected.
To confirm the gene expression profile determined by microarray, several selected genes were subjected to RT–PCR analysis, using total RNAs derived from the two independent samples of LEC, MC-L, and MSC-BM that were used for the microarray experiments, as well as an additional pair of LEC, MC-L, and MSC-BM samples. Ribosomal protein large 35 (RPL35) a, a ribosomal protein served as an internal control. A 2 µg quantity of RNA was reverse transcribed using a cDNA synthesis kit (Applied Biosystems, Foster City, CA) and 1/100th of the reaction was used per 20 µl PCR reaction. PCR reactions were performed with DyNAZYME master mix (Finnzymes Oy, Espoo, Finland). The PCR products were resolved on a 2% agarose gel containing ethidium bromide. Real-Time PCR quantitation was performed in an ABI prism 7900 HT sequence detection system and analyzed with SDS 2.1 software (Applied Biosystems). The reactions were identical to those described above, except that DyNAMOTMSYBERgreen 2× mix (Finnzymes Oy) was used in place of DyNAZYME MIX. The sequences of primers are shown in Table 1. Amplification of RPL35a was performed for each cDNA (in triplicate) for normalization of RNA content. Threshhold cycle number (Ct) of amplification in each sample was determined by ABI Prism Sequence Detection System software (Applied Biosystems). Relative mRNA abundance was calculated as the average for Ct for amplification of a gene-specific cDNA minus the average Ct for RPL35a and fold change over control has been calculated as follows:
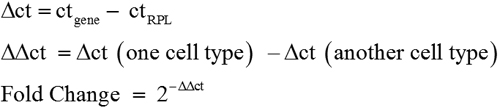
Three individual gene-specific values thus calculated were averaged to mean±standard deviation, and fold change was expressed as log 2 ratios.
Spindle cell cultures were established from extended limbal explant cultures after 2 to 3 weeks of culture. Under a phase contrast microscope the cells appeared fibroblastic, elongated, and spindle shaped and few cells were large and flat with a single nucleus. These cells demonstrated the ability to form colonies with the occasional cell sphere formation giving the impression of embryoid bodies (Figure 1).
The LEC on immunocytochemical analysis, showed immunoreactivity toward ABCG2, CK3/CK12, CK14, PAX-6, CDH1, and vimentin. MC-L were found to be immunoreactive for vimentin and nestin and were negative cytokeratin 3/12 (KRT3/12), cytokeratin 14 (KRT14), and CD45 (Figure 2A-L).
Flowcytometry analysis of MC-L revealed expression of CD90+CD44 (Figure 2N [Q2=98.4%]), CD13 (Figure 2V [Q1–3=86.4%]), and HLA-ABC (Figure 2P [Q1=91.1%]) and were negative or weak expression for CD34 (Figure 2Q [Q4=0.0%]), CD10 (Figure 2Q [Q1=0.1%), CD34+CD10 (Figure 2Q [Q2=0.0%]) CD11b (Figure 2P [Q4=0.0%]), CD40 (Figure 2O [Q4=0.0%]), CD40L (Figure 2T [Q4–1=0.0%]), CD138 (Figure 2V [Q4–3=0.0%]), CXCR4 (Figure 2S [Q4–1=0.0%]) and HLA-DR (Figure 2O [Q1=0.1%]) expression (Figure 2M-V).
In this study two different samples of LEC, MC-L, and MSC-BM were labeled using (Cy3 and Cy5) dyes and competitive hybridization was performed. The data has been deposited in NCBI’s Gene Expression Omnibus (GEO) with GEO series accession number (GSE16763). The fold change was expressed as average of two different biologic samples. Analysis of the data considering a threefold difference, suggested differential expression of 3,484 genes between LEC and MC-L; 1,579 genes between MC-L, and MSC-BM and 4,837 between LEC and MSC-BM. The differentially/highly expressed genes in LEC, MC-L, and MSC-BM are shown in Table 2. The groups were segregated based on the average fold expression toward one lineage as compared to other, i.e genes that are highly representative of: a) LEC≥25 fold expression as compared to MC-L and MSC-BM; b) MC-L≥15 fold expression as compared to LEC and MSC-BM; c) MSC-BM≥20 fold expression as compared to LEC and MC-L; d) MC-L and MSC-BM≥20 fold expression compared to LEC; e) LEC and MC-L≥10 expression compared to MSC-BM.
Some of the highly expressed genes in LEC include the CD24 (48 and 155 fold overexpression compared to MSC-BM and MC-L, respectively), FOXA1 (27 and 631 fold overexpression compared to MSC-BM and MC-L, respectively) and KRT13 (11 and 15-fold), LAMA3 (7.4 and 7.3), ITGA6 (22 and 10) and CDH3 (6.0 and 7.4). To explore the interdependence of LEC and MC-L, we looked at the growth factor and cytokine profile of these cells (Table 3). LEC showed high expression of growth factors like transforming growth factor alpha (TGF−α), Amphiregulin (AREG), epiregulin (EREG), hepatocyte binding epidermal growth factor (HB-EGF), growth factor receptor-bound protein 14 (GRB14), fibroblast growth factor 11 (FGF11), and cytokines like chemokine (C-X-C motif) ligand 1 (CXCL1), CXCL2. The MC-L showed high expression of growth factors like FGF7, FGF2 and cytokine CXCL12.
The analysis of MC-L and MSC-BM showed similar and differential gene expression between the two cells. Various gene ontology terms were picked up and analyzed from the microarray data. The gene ontology terms were classified into groups like osteogenic, chondrogenic, myoblast, adipogenic, MHC-class II related, Homeobox genes, extracellular, and other genes (Table 4).
To validate the gene expression profiles determined by the microarray analysis, the expression levels of selected genes were analyzed by real time PCR (10 genes; Figure 3) and semi quantitative RT–PCR (28 genes, Figure 4). The gene expression patterns obtained by the two techniques were in good agreement with that from the microarray analysis, indicating high fidelity in microarray data and analytical methods.
Limbal stem cell deficiency has been a challenging clinical problem, the current treatment of which involves replenishing the depleted limbal stem cell pool by either limbal tissue transplantation or use of cultivated limbal epithelial sheets [1-3,24]. As described in our earlier publications [25,26], we established a feeder cell free method of cultivating the limbal explant tissues on denuded human amniotic membrane. Our results show that limbal explant culture derived MC-L when expanded exhibit a spindle shaped, fibroblast-like appearance similar to that of MSC-BM [17]. Though we had no logical explanation for this in the beginning, the revelation of presence of spindle cells prompted us to postulate that these spindle cells in the explant culture system function like “intrinsic feeder cells.” To the best of our knowledge and literature search this is the first study that characterizes the cultured cells (LEC as well as MC-L) from the limbal explant culture, which are directly used for translational research in humans. Isolated MC-L can be distinguished from epithelial cells (lack of expression of KRT3/12, KRT14), fibroblasts (lack of expression of HLA-DR), hematopoietic stem cells (lack of expression of CD34, CD45, CD11b, CD10, CD40, CD40L, and CD138), because they are adherent to the surface of tissue culture flasks and express different cell-surface markers (CD90, CD13, CD105, and CD44).
Genes that show differential expression in the LEC when compared to MC-L and MSC-BM, encode proteins that stabilize epithelial sheets and promote or regulate cell to cell interaction and cell to matrix interaction including keratins (Keratin 13, Keratin 12), laminins (LAMA3, LAMB3), cadherins (CDH3 and CDH1), nebulette, epiregulin, calbindin 1 28 kDa, desmosomal components (DSG3, DSC2), matrix metallo peptidase 10, Serine peptidase inhibitor clade B5, and carcinoembryonic antigen-related cell adhesion molecule 1. In addition, LEC showed high expression of known basal markers (TP73L [p63], ITGA6, epiregulin, and HOP) and differentiated epithelial markers (CDH1, KRT12) [27]. Immunocytochemical analysis showed the expression of limbal epithelial stem cells markers ABCG2, vimentin, KRT14, and KRT19 and also expressed the differentiated epithelial markers CDH1 and KRT3/12 on cultivated LEC. This further supports the fact that cultivated LEC cells on dhAM in a feeder cell- free culture technique, contain a distinct population of stem cells and differentiated cells which serve to replenish the depleted limbal stem cells when transplanted to the diseased eye [1,2,25]. Some of the high expressed transcripts in the limbal epithelial cell cultures include CD24, a surface molecule that has been used to identify different types of human stem cells [28]; OTX1, a transcription factor is expressed in the presumptive ciliary body and iris and has been shown to be essential for development of these tissues [29], and FOXA1, an endodermal stem cell marker [30]. While it could be critically argued that the study does not represents the specific signature of the limbal stem cells alone, this data are valuable in contribution to data on entire population of cultured limbal epithelial cells that are used for clinical transplantation. This further supports strong evidence that the cultivated LEC cells contain a distinct population of stem cells and differentiated cells which serve to replenish the depleted limbal stem cells when transplanted to the diseased eye – it represents the signature of cultured limbal cells inclusive and not exclusive of stem cells.
Cytokine and growth factor signaling is an important determinant of the functional state of these cells and therefore we evaluated this relationship between LEC and MC-L [15]. The most important growth factors for normal human keratinocyte proliferation are member of EGF family, including TGFA, HB-EGF, ER, and AR and these act in an autocrine manner [31]. Our data also reveals the high expression of these four EGF members in LEC. The fibroblast growth factors, FGF1 and FGF2, are well characterized growth factors known for their mitogenic effect on several cells derived from neuroectodermal or mesodermal origins. FGF1 and FGF2 (mitogenic to corneal and limbal epithelium) [32], keratinocyte growth factor (FGF7), epithelium specific growth factor (mitogen for several epithelial cells including limbal epithelial cells) [15] were highly expressed in MC-L. Interestingly, and as expected, their corresponding receptors FGFR1 were expressed in MC-L and FGFR2 in LEC. The proinflammatory forms of IL-1 (IL1A and IL1B) expression in LEC play significant roles in ocular surface immune and inflammatory responses and wound healing [33]. The highly expressed chemokine observed in LEC include CXCL1, 2, 3, 10, and 11 and in MC-L include CXCL12, CCL26, CCL2, and CCL13. The intense expression of chemokine ligand CXCL12 (Stromal cell derived factor 1) in MC-L is similar to the study by Tristan and coworkers [34]. This factor might exert physiologic effects on the cornea and could be involved in pathological conditions such as corneal angiogenesis [34]. The neurotrophic factors have been reported to play important roles in maintaining stem cells in the limbus [35]. We also noted a high expression of neurotrophic factors like neurotrophin 3, nerve growth factor and brain derived growth factor in MC-L while neurotrophin 5 was highly expressed in LEC. Glial derived neurotrophic factor (GDNF) and its receptor GDNF receptor alpha 1 were highly expressed in the MC-L and MSC-BM similar to the observations made by Qi and coworkers in limbal cells [35]. All these features support our hypothesis that the limbal epithelial cells and stromal cells play a complementary role not only in vivo but also in vitro in the explants culture system.
Genes highly expressed (Table 2) in MC-L include fetal kidney cadherin (CDH6), vascular endothelial growth factor receptor 1 (VEGFR1 or FLT1) glutamate receptor ionotrophic (GRIA3), collectin subfamily member 12, transcription factor forkhead box F1 (Foxf1), src homology 2 domain contataining transforming 3 (SHC3), oxytocin receptor, and unknown genes AFO52115 and BCO73929. These genes with such higher expression (>15 fold) can be considered as the markers of mesenchymal like cells of limbus. The FOXF1 is a transcription factor which is expressed in mesenchymal cells of the lung, liver and gall bladder and is shown to be involved in mesenchymal cell migration without changes in cell proliferation and cells survival [36].
An interesting observation was the high expression of receptors neurophilin 1, platelet derived growth factor receptor alpha, and leprecan-like 2 in MC-L, which is similar to MSC-BM [37]. This study supports the characteristics of mesenchymal cells (MSC-BM, and MC-L) that were previously identified in MSCs, such as vimentin, fibronectin, collagen Type I and III, collagen type VI, light chain of myosin 9, and matrix metallopeptidase 2 [37-40]. The genes which show similar gene expression in MC-L and MSC-BM are those which are related to extracellular components, cell adhesion molecules (microfibrillar associated protein 5, syndecan 2, matrix-remodelling associated 5, chondroitin sulfate proteoglycan 4, and collagen 8 alpha 1) and the genes related to osteoblasts (osteonectin, collagen type 1, connective tissue growth factor, and OB-cadherin), chondrocytes (fibromodulin, decorin, tensin 1, hyaluronan, and proteoglycan link protein) and myoblasts (transgelin, sarcoglycan epsilon, caldesmon 1, leimodin, and meltrin alpha; Table 4). The MC-L also expressed the products characteristics of hematopoiesis-supporting stroma, including fibulin-1, fibulin 2, collagen type VI, and stromal cell-derived factor, in the same level as MSC-BM thus supporting our hypothesis that these cells possibly act as intrinsic feeder cells or nurture cells in explant culture system. Nevertheless, some differences were observed between expression profiles of MC-L and MSC-BM. Among the genes that were exclusively or expressed at higher levels by MSC-BM are growth differentiation factor 6, neurotrophic tyrosine kinase receptor 2, urea transporter erythrocyte, and myogenic factor 6. Other genes highly expressed at higher levels in MSC-BM include chondrogenesis related genes (cartilage oligomeric matrix protein, collagen 11 alpha 1, and chitinase 3 like 1), osteogenic related genes (runt related transcription factor 2 and osteopontin) and adipogenic related genes (CCAAT/enhancer binding protein alpha, leptin, and Serum amyloid A1 transcript variant 1). The MSC-BM are more committed to the osteoblastic, chondrogenic and adipocytic lineages. This suggests that in addition to some common signatures of niche supporting cells, mesenchymal cells from different sources possibly carry tissue specific signatures, which reflect their tissue of origin. The limitations of study would be the sample size used for microarray (n=2) and potential contamination of stromal cells in limbal epithelial cells cultures.
In summary, this study highlights the gene expression profile of cultivated limbal epithelial cells, mesenchymal cells from limbal stroma obtained from an ex-vivo expanded, feeder cell-free, limbal explant tissue culture system. Their lineage specific signatures, evidence of interdependent pathways with limbal epithelial cells, striking resemblance to the signature of bone marrow derived mesenchymal cells support our hypothesis that the limbal stromal cells act like intrinsic feeder cells or the nurture cells, similar to bone marrow derived mesenchymal cells and could possibly be an important component of limbal niche.
The Council for Scientific and Industrial Research is acknowledged for providing a research fellowship to Naresh Polisetti. Prof. Paturu Kondaih’s laboratory is supported by infrastructural grants to MRDG from University Grants Commission (SAP program), Department of Biotechnology (Program support) and Department of Science and Technology (FIST) program. Further support was from The Hyderabad Eye Research Foundation, Department of Biotechnology, India, Sudhakar and Sreekanth Ravi Stem Cell Biology Laboratory, and the Antonio Champalimaud Foundation.