Received 14 March 2003 | Accepted 27 August 2003 | Published 5 September 2003
Reprint
Received 14 March 2003 | Accepted 27 August 2003 | Published 5 September 2003 |
Download Reprint |
The dual role of dexamethasone on anti-inflammation and outflow resistance demonstrated in cultured human trabecular meshwork cells
Yuk Fai Leung, Pancy Oi Sin
Tam, Wing Shan Lee, Dennis Shun Chiu Lam, Hin Fai Yam, Bao Jian Fan,
Clement Chee Yung Tham, John Kien Han Chua,
Chi Pui Pang
(The first two authors
contributed equally to this publication)
Department of Ophthalmology & Visual Sciences, The Chinese University of Hong Kong, Hong Kong
Correspondence to: Prof. C. P. Pang, Department of Ophthalmology &
Visual Sciences, The Chinese University of Hong Kong, Hong Kong Eye
Hospital, 147K Argyle Street, Kowloon, Hong Kong; Phone:
+852 27623169; FAX: +852 27159490; email: cppang@cuhk.edu.hk
Dr. Leung is now at the Bauer Center for Genomics Research, 7 Divinity
Avenue, Harvard University, Cambridge, MA.
Abstract
Purpose: Dexamethasone (DEX) is a glucocorticoid commonly used in topical eyedrops to treat eye inflammation. It has an undesirable effect of inducing glaucoma in certain patients. In human Trabecular Meshwork (TM) cells DEX regulates a number of genes but its global influence on TM gene expression is still elusive. In the present work, DEX effects on global gene expressions of an established human TM cell line were studied by microarray.
Methods: The whole experiment of microarray was repeated three times. Differentially expressed genes were identified by an empirical Bayes approach and confirmed by Reverse Transcription Polymerase Chain Reaction.
Results: Eight genes (GAS1, CDH4, MT1L, CST3, ATF4, ASNS/TS11, CHOP, HSPA5) were identified that are at least a thousand times more likely to be differentially expressed due to DEX treatment and six genes (TSC22, LDHA, IGFBP2, TAGLN, SCG2, WARS) were identified that are at least a hundred times more likely to be differentially expressed due to DEX treatment. Except for MT1L, ASNS/TS11, IGFBP2, SCG2, and WARS, all the other genes are first reported here to be regulated by DEX in TM. Intriguingly, several of them have overlapping roles in anti-inflammatory response and outflow resistance.
Conclusions: The results of our experiments on cultured human TM cells indicate that the increase in outflow resistance and ultimate ocular hypertension may be byproducts of the favorable anti-inflammatory response triggered by DEX.
Introduction
For a long time glucocorticoid, commonly dexamethasone (DEX) in topical eye drops, has been used to treat eye inflammation, which is characterized by vasodilation, increased vascular permeability, and cellular infiltration and activation. These processes are regulated by a number of internal and external factors including chemokines and cytokines. In the inflammation process, usually extensive extracellular matrix (ECM) remodeling is involved. At least part of the anti-inflammatory effect is mediated through its inhibitory actions on cytokines, receptor synthesis, [1] and neurophil activation [2]. However, glucocorticoid treatment also causes undesirable side effects of inducing ocular hypertension or glaucoma [3,4]. Although the exact mechanism is not completely understood, numerous in vivo and in vitro studies have indicated that long term treatment of DEX on trabecular meshwork (TM) can induce drastic changes in extracellular matrix protein expression and turnover, cytoskeletal structure, cell adhesion, morphology, and function [5]. These biological changes have considerable implications on glaucoma pathogenesis and therapy.
The prolonged effects of DEX on human TM cells provided the fundamental data to clone the first glaucoma gene, myocillin (MYOC) [6-9]. At least 50 mutations in MYOC have been shown to associate with juvenile and late-onset primary open angle glaucoma (POAG) to-date. Intriguingly, MYOC mutations are present only in a minor proportion, about 4.6%, of POAG patients [10].The mutation pattern appears to be population-specific. We have identified a different MYOC sequence alteration pattern in Chinese people when compared to Caucasian [11,12] people. The candidate gene for the GLC1E locus [13] was recently identified as optineurin (OPTN) [14]. Mutations were found in 16.7% of hereditary POAG families. Our preliminary screening of OPTN coding exons identified a different mutation pattern in the Chinese population again (data not shown). Among other potential POAG loci, such as GLC1B-D and -F, specific genes are still to be identified [15-17]. Obviously the etiology and pathogenesis of POAG are complex.
The paradigm of life science research is shifting from gene-by-gene basis to a global view of gene interaction networks. Microarray technology is one of the most promising technologies in this area. Since its first introduction in 1995 [18], microarray has been shown to be useful in expression profiling [19], disease classification [20,21], and pathway reconstruction [22]. Microarray expression profiling is one of the most promising methods for identifying new glaucoma candidate genes, which may be diagnostic biomarkers or even targets for novel therapeutics development. However, eye functional genomics study by microarray is still in its infancy, and published reports are emerging rapidly. One of the examples was a study by Gonzalez et al. who used a membrane microarray to investigate the effect of elevated intraocular pressure (IOP) on human TM and identified specific upregulation of 11 genes [23]. Recently Ishibashi et al. isolated primary TM cell cultures from normal human cadaver eye and performed microarray analysis on their expression profile changes after DEX treatment [24]. Interesting results on differentially expression genes under DEX treatment were obtained. However, there was no information about the steroid responsiveness about the eye donors. Besides, the microarray experiments in both studies [23,24] were not performed in replicate, thus it is difficult to evaluate the statistical significance of differentially expression genes.
In this study a microarray with 2400 genes was used to investigate differential gene expressions of an established human TM cell line under DEX treatment and carry out initial characterization of the differentially expressed genes. Since the prolonged effects of DEX on this TM cell line provided the fundamental data to MYOC [6-9], this cell line is the ideal candidate for studying the DEX response on TM. The microarray experiment was repeated three times [25] and the differential expression was inferred by an empirical Bayes method [26]. The results were confirmed by reverse transcription-polymerase chain reaction (RT-PCR). A total of nine genes were shown to be at least a thousand times more likely to be differentially expressed, among them four up-regulated and five down-regulated. Another six genes were at least a hundred times more likely to be differentially expressed, four of them up-regulated and two down-regulated. Most of them were not previously identified as DEX regulated genes in TM. These genes share substantial functional and pathway overlap on anti-inflammatory response and outflow resistance. The current results on cultured human TM cells indicated that steroid induced outflow resistance and subsequent ocular hypertension are by-products of the favorable anti-inflammatory response imposed by glucocorticoid treatment.
Methods
Cell Culture and DEX treatment
TM cell line in the eighth passage was used for DEX treatment. The cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (Invitrogen Life Technologies, Carlsbad, CA, USA), maintained until confluent before DEX (water-soluble dexamethasone, Sigma-Aldrich Co., St. Louis, MO, USA) treatment. Effects of DEX on gene expression of TM cells was tested by adding 100 nM DEX or water as control to the medium. The amount of DEX added to the medium corresponds to the level achieved in the aqueous humour by topical corticosteroid eyedrops [9]. The cells were then maintained in DEX or water control treatment for a total of 10 days before harvesting. During DEX treatment, the medium was changed every other day. After DEX treatment, the cells in full confluence did not show fine morphology changes under light microscopy, although some increase in cell and nuclear sizes were noted. Three individual sets of DEX treated and control cells were prepared for each microarray replicate.
RNA extraction
All cells were harvested on the tenth day of DEX treatment. The cells were rinsed three times in phosphate buffer saline, centrifuged, lysed in RLT buffer (Qiagen, Hilden, Germany) with 10 μl/ml β-mercaptoethanol and homogenized by QIA shredder column (Qiagen). Total RNA was extracted by phenol-chloroform [27] and then by RNeasy kit (Qiagen). The final RNA concentration was quantified on a GeneQuant RNA/DNA calculator (Amersham, Uppsala, Sweden) and the quality affirmed by agarose gel electrophoresis.
Microarray experiment
MicroMax Human cDNA System I (PerkinElmer Life Sciences, Boston, MA, USA) with 2400 genes was used in the microarray experiment. The same lot of microarrays (chip lot: 146408) was used for all three replications. Twelve plant genes were spotted on the microarray and exogenous plant RNA was added as an internal control. Therefore the actual number of human genes on the array was 2388. The RNA from equal number of cells from DEX treatment and control were labeled with Cy3 and Cy5, respectively by the MicroMax Direct Reagent kit, hybridized to the microarray overnight at 65 °C, washed and dried according to the manual from the supplier.
Data analysis
The fluorescent images were scanned by a ScanArray 4000 scanner (Parkard Biochip Technologies, Billerica, MA, USA). The images were visually inspected to flag and exclude the abnormal spots with irregular shape or dirt. The internal plant control genes were also excluded.
Only the spots with signals higher than background by two standard deviations were used for subsequent analysis. All image acquisition and raw signal intensity extraction were performed by ScanArray version 2.1 and QuantArray version 2.0, respectively (Packard Biochip Technologies, Billerica, MA, USA). The signal on each slide was normalized using within-print-tip-group lowess normalization with default parameters [28]. Spatial plots and boxplots were used to evaluate whether the within-print-tip-group lowess normalized data required any scaling. Then the log intensity ratios were normalized across the slides so that each slide had the same median absolute deviation. The expression level of genes is denoted as Mij,

where i is the number of genes on the array (1...N) and j is the number of replications (1...n). In this experiment N=2400 and n=3.
Assuming the Mij has a Gaussian distribution with mean μi, the log posterior odds for each gene to be differentially expressed were calculated using a Bayes method,
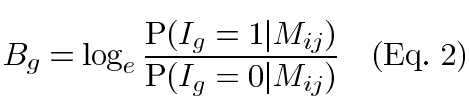
where Bg is the log posterior odds for gene g to be differentially expressed, Ig=1 indicates the gene g is differentially expressed and Ig=0 indicates the gene g is unchanged.
The formula for Bg is
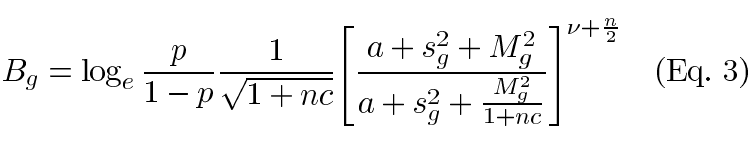
where p is the prior distribution of differentially expressed genes. In this study, Bg was set at the default value 0.01. sg2 is the sum of square (SSB) over replications, a and ν are hyperparameters in the inverse gamma prior distributions of the variances; c is the hyperparameter in the normal prior of the nonzero mean. Derivation and usage of this formula was detailed by Lee et al. [25]. All spatial plots, normalization, and Bayesian analyses were carried out by the R package "Statistics for Microarray Analysis" (SMA) version 0.5.7 implemented in the R environment (version 1.4.0). All other analyses were performed in the R environment (version 1.4.0).
RT-PCR
Differential expression of those genes with B statistics >=2 was verified by semi-quantitative RT-PCR. The differentially expressed genes were compared from DEX-treated and control cells using primers specially designed to anneal with the sequences located in different exons. RT-PCR was performed at the exponential phase with the number of cycles determined for each tested gene. Three housekeeping genes, beta-actin (ACTB), glyceraldehyde-3-phosphate dehydrogenase (GAPD), and pyruvate dehydrogenase beta-subunit (PDH), were used as internal controls. The experiments were repeated three times for verification. In each RT-PCR 1 μg of the total RNA was reverse-transcribed with Superscript II reverse transcriptase into cDNA, and 10 ng was used as template for PCR amplification, with primers and PCR conditions depicted in Table 1. Specific PCR cycle conditions were selected within the range of the linear amplification for each gene to reflect the level of differential expression. The size of each amplicon, which was the RT-PCR product (Table 1), and reaction specificity were confirmed by agarose gel electrophoresis. The gel image was captured and expression ratios calculated by Bio-Rad Gel-Doc 2000 System (Bio-Rad Laboratories, Hercules, CA, USA) and Quantity One® version 4.0.3 (Bio-Rad), respectively.
Results
Microarray analysis
Among the 2400 genes on the MicroMax microarray, 12 were exogenous non-mammalian control genes. The spots with abnormal shape, dirt, or signal lower than local background intensity plus two standard deviations were flagged and ignored in subsequent analysis. A total number of 7139 data points, corresponding to raw data of 2388 human genes in three replicates, were used for subsequent analysis (Table 2). The raw data was input to SMA for further analysis. Print-tip group lowess normalization was chosen for normalization for all three slides. Multiple slide normalization was then performed across the three slides, allowing their log-ratios to have the same median absolute deviation. A log posterior odds, B statistics, was calculated using this dataset, the resulting B vs M plot shown in Figure 1. A total of nine genes, as denoted by red crosses, had B statistics greater than or equal to 3. This means they were at least a thousand times more likely to be differentially expressed than to remain unaffected. Among these nine genes, four were up-regulated: growth arrest-specific 1 (GAS1), cadherin-4, (CDH4), metallothionein 1L (MT1L), and cystatin C (CST3). Five were down-regulated: activating transcription factor 4 (ATF4), asparagine synthetase (ASNS), hybrid gene of RNA-binding protein FUS and C/EBP-homologous protein (TLS/CHOP), heat shock 70 kD protein 5 (HSPA5), and the ts11 gene encoding a G-1 progression protein (TS11). Another six genes, as denoted by blue asterisks, had B statistics greater than or equal to 2 and less than 3, meaning they are at least a hundred times more likely to be differentially expressed. Among them, four were up-regulated: transforming growth factor beta-stimulated protein TSC-22 (TSC22), lactate dehydrogenase A (LDHA), insulin-like growth factor binding protein 2 (IGFBP2), and transgelin (TAGLN). Two were down-regulated: secretogranin II (SCG2), and tryptophanyl-tRNA synthetase (WARS) (Table 3).
Confirmation of differential gene expression by RT-PCR
The differential expression of those genes with B >=2, together with three housekeeping genes (GAPD, ACTB and PDH) and MYOC, were confirmed by RT-PCR (Table 1). In general the microarray and RT-PCR expression ratios matched well (Figure 2, Table 3) while the three house keeping genes had similar expression ratios in both DEX-treated and control cells. ASNS and TS11 were found to have exactly the same sequence during the primer design stage. It was revealed that their earlier annotations in GenBank were completely different but now they are treated as the same gene or synonym in the curated databases. They probably were regarded as different genes when incorporated in the array during the design process. Therefore, RT-PCR was only performed for one of them. S62138 is a hybrid gene consisting of a TLS and CHOP domain. Since this hybrid protein is a product of a characteristic chromosomal translocation occurred in human myxoid liposarcoma [29], it is not likely to be present in a normal TM cell culture, whether or not treated with DEX. This was also confirmed by RT-PCR using a primer set spanning TLS and CHOP. A more realistic scenario was that both the TLS and CHOP genes were bound to the hybrid gene on the array and gave a mixed signal. As a result, RT-PCR reactions were performed to confirm the differential expressions of TLS and CHOP.
Although MYOC was included in the array design, the data analysis did not reveal any significant differential expression as would be expected. Careful inspection of the microarray data revealed consistent manufacturing defects in the same batch of the array and MYOC was inside one of them (Figure 3). The genes within the affected areas were flagged and excluded from further data analysis. Therefore, there was no conclusive data about MYOC from our array results. Since MYOC is well documented to be up-regulated by DEX treatment, RT-PCR was used to affirm it expression. We found MYOC up-regulated by 4.32 fold (Table 3).
Correlating the gene functions by referencing to knowledge base
In order to further understand the functions of these genes, the available Gene Ontology terms and SWISS-PROT functions/pathways and subcellular annotations were extracted and listed in Table 4.
Discussion
The paradigm of life science research has been changing from one-by-one genetics to a holistic understanding of biological systems. Microarray is one of the most powerful tools for this purpose. It not only allows a simultaneous inspection of thousands of gene expression levels in different samples, but also makes possible the study of global gene functional networks when accompanied with appropriate experimental design and data analysis [30]. This ultimately contributes to a better understanding of complex disease pathogenesis. We used microarray in this study to investigate the global effects of DEX on gene expression of cultured human TM cells.
Researchers often do not repeat microarray experiments due to high cost and difficulties to obtain sufficient amounts of RNA. Differential expression is usually inferred if a gene's expression ratio is greater than 2 or less than 0.5. Sometimes RT-PCR is performed in parallel to confirm or support the detected changes of expression levels. It has been shown that replication of microarray experiments is beneficial to understanding the underlying variance of the data. This also provides a means to identify statistically significant differential expressions and considerably reduce false positives [25]. Therefore, in this study the whole set of experiments starting from the cell culture stage was repeated for three times.
This study has revealed a number of possible problems that virtually everyone would encounter when using commercial microarrays. The first one is limited quality check. Although the overall quality of MicroMax is very high, yet a small manufacturing defect in the same batch of array has affected our data collection on the MYOC gene (Figure 3). This type of defect is generally not a problem for researchers who produce their own arrays because some arrays can be sampled to perform mock hybridization for quality control. Defective arrays can be discarded. However this quality check is not possible when using expensive commercial arrays. Replications cannot solve this particular problem because the defects occurred in the same region of the same batch of arrays and, subsequently, all data for the same spot are lost. This problem is partly solved by the newer version of MicroMax with genes arrayed in duplicate in different areas on the same slide. At least part of the data can be recovered by the duplicated spots. The other problem is clone selection strategy. The users have limited control over the commercial array design, while a great deal of bioinformatic analysis is necessary to select the appropriate genes or appropriate part of the genes to make up the array [31]. In this experiment there was a gene spotted twice on the array, ASNS and TS11 (Table 3, Table 4), possibly because they occurred in GenBank with different annotations. The ASNS has been defined as human asparagine synthetase mRNA and TS11 human ts11 gene encoding a G-1 progression protein, whilst they are referred to the same gene in curated databases like SWISS-PROT. A better cross-checking during the design stage can avoid this problem. Another problem observed in this experiment was the presence of a hybrid gene TLS-CHOP on the array. This is a product of chromosome translocation in human myxoid liposarcoma. It is not likely that such a gene product would have been detectable in the current experimental system or POAG. A subsequent RT-PCR confirmed that the TLS-CHOP hybrid gene was not expressed in our system. A careful array design can avoid this confusion. Nonetheless, duplication of ASNS and TS11 provided an empirical measure for our microarray hybridization precision. The mean expression ratio of ASNS and TS11 obtained from the array was 0.483 and 0.421, respectively. They were nearly identical and coherent with a RT-PCR expression ratio of 0.415.
A superior solution to all these problems is to use oligonucleotides arrays instead of cDNA arrays, either home-made arrays or commercial arrays like Affymetrix. In oligonucleotide microarrays, different regions of the same gene can be represented by different oligonucleotide spots. This type of probe redundancy can greatly increase the reliability of differential expression results. Good oligonucleotide arrays safeguard the quality of the collected data. Although the initial effort and cost to make or use oligonucleotide arrays appear to be much greater than those of cDNA arrays, this is not necessarily the case with the ultimate expenses. There are hidden costs in making cDNA arrays like sequence verification, clone purchasing, and labor. As a result, serious microarray researchers are exploring oligonucleotides arrays for routine use.
In the present study, RT-PCR analysis confirmed differential regulation of genes with B greater than 2 (Table 3, Figure 2). As mentioned earlier, the MYOC data collection was affected by a manufacturing defect, therefore its differential expression was affirmed only by RT-PCR, which revealed up-regulation of the MYOC gene in the DEX treated cells by 4.32 fold. This is consistent with other studies on MYOC regulation by long-term DEX treatment [6-9]. Since the functional properties of differentially expressed genes may provide clues to understand the biological effects of DEX treatment on TM cells and hence glaucoma pathogenesis, the Gene Ontology terms and SWISS-PROT function/pathways and subcellular location annotations of those genes with B greater than 2 were extracted from respective databases (Table 4). A number of notable findings related to possible DEX effects on anti-inflammation and outflow resistance were identified from their possible functions. MYOC was up-regulated by 4.32 fold. This is not surprising because MYOC was originally identified by DEX induction on TM cells. MYOC is both secreted and expressed intra-cellularly [6]. Extra-cellular MYOC preferentially associates with fibronectin [32] and co-localizes with type IV collagen [33]. The induced extracellular MYOC likely associates with and stabilizes the extracellular matrix (ECM) and ultimately increases the outflow resistance. Intracellular MYOC is associated with sub-cellular structures like endoplasmic reticulum (ER) [34] and Golgi apparatus [35], as well as co-localizes with microtubules [36] and mitochrondria [37]. Disruption of TM cytoskeleton could increase outflow facility [38].
GAS1 was up-regulated by 3.28 fold. It is an integral membrane protein and suppresses cell proliferation by blocking entry to S phase [39]. It has been shown to be induced by DEX in NIH-3T3 fibroblasts [40]. The presence of an extracellular arginine-glycine-aspartic acid (RGD) sequence suggests GAS1 may interact with integrins and modify the attachment of cells to the ECM [40]. Therefore its up-regulation might moderate the cell proliferation induced by inflammation but at the same time increase cell-cell and cell-ECM adhesion and hence compromise cell permeability. This proposition is consistent with the 2.85 fold up-regulation of CDH4 detected in this study. CDH4 was originally cloned from brain and retina cDNA libraries [41]. It is strongly induced by DEX in human osteoblastic cells [42] and has a potential role in striated muscle formation [43]. Cadherin plays a role in cell-cell adhesion in bovine aortic endothelial cells (BAEC). Latrunculin-A, a drug used to destroy cadherin interaction in BAEC, was found to increase the outflow facility two to three fold in monkeys [44]. It was believed that the destruction of cell-cell adhesion junctions and cell-ECM focal contacts in juxtacanalicular and corneoscleral regions would render the overall meshwork architecture less rigid. Over-production of cadherin, on the other hand, might produce an opposite effect that induces abnormal adhesion and decreases the aqueous outflow facility.
Normal TM expresses a number of ECM proteins. For example the amorphous basement membrane-like material in juxtacanalicular tissue consists of mainly collagen type IV, laminin and fibronectin. In sheath-derived plaques many ECM proteins including elastin, fibrillin-1, microfibril-associated glycoprotein-1, decorin, and collagen type VI have also been detected [5]. MYOC was shown to interact with fibronectin and fibrillin-1 [45]. In glaucoma with increased outflow resistance, there was abnormal accumulation of these ECM proteins in TM [46,47]. DEX was able to induce some of these ECM proteins including collagen IV [48], fibronectin [49] and elastin [50] in TM. Although such DEX associated ECM proteins were not on the array in this study, we noticed an interesting finding. The CST3 was up-regulated by 1.95 fold. CST3 is the most abundant extracellular inhibitor of cysteine proteases. Common cysteine proteases like cathepsins B and L are usually up-regulated in tumor progression, metastasis, tissue injury and inflammation [51]. Cathepsin L was also up-regulated by elevated IOP in TM in an earlier microarray study [23]. Such up-regulation can help the degradation of ECM including major components in TM, for example collagen type IV, laminin and fibronectin [52,53]. This can increase vascular permeability and facilitate cellular infiltration. Therefore one of the anti-inflammatory effects of DEX is likely to be mediated via the up-regulation of the cysteine proteases inhibitor CST3. The DEX induction of CST3 has also been demonstrated in HeLa cells [54]. Unfortunately, this anti-inflammatory up-regulation of CST3 also comes with a price. The decrease in ECM turnover due to its inhibitory activities against cysteine proteases potentially leads to an increase in outflow resistance.
Besides the extracellular changes, DEX treatment also induces a drastic reorganization of the cytoskeleton into cross-linked actin networks (CLANs), which resemble geodesic dome-like polygonal lattices [55]. CLANs were also identified in a number of TM cell lines derived from glaucomatous donors, suggesting their possible roles in POAG. In our experiment TAGLN, was up-regulated by 1.53 fold. TAGLN is an actin cross-linking and gelling protein and is possibly involved in the production of CLANs. While disruption of the TM cytoskeleton could increase outflow facility [38], the abnormal accumulation of CLANs, in contrast, would stabilize the TM cytoskeleton and cell-matrix attachment [5]. Together with the up-regulation of CDH4, decrease in ECM turnover and increase in ECM production and stability, the efficiency of aqueous outflow can be greatly compromised. At the same time, the tighter cell-cell and cell-matrix adhesion induced by DEX might reduce cellular infiltration, which is a typical inflammatory response [56].
The 1.79 fold up-regulation of TSC-22 potentially contributes to aqueous outflow. TSC-22 was identified as a leucine zipper containing protein induced by transforming growth factor beta 1 (TGFβ-1) and up-regulated by DEX in mouse osteoblastic cells [57,58]. It activates C-type natriuretic peptide (CNP) expression [59]. In rabbit eyes CNP has an ocular hypoptensive effect through increasing the outflow facility when injected intravitreally [60]. However, this DEX-induced hypotension appears contradictory to IOP elevating effect of TSC-22. Depending on the dimerization partner, TSC-22 can act either as transcription repressor or activator [58]. The SWISS-PROT annotation for TSC-22 is transcriptional repressor. In fact, the transcription regulation mechanism of TSC-22 is still not completely clear. It is possible that TSC-22 preferentially binds to its transcription repressor partner and inhibits CNP expression in TM under DEX treatment. CNP repression can ultimately lead to increased IOP.
ATF4 and CHOP were down-regulated by 1.81 and 1.25 fold, respectively, as revealed by RT-PCR. ATF4 is also known as cAMP-responsible element binding proteins (CREB) that regulates expressions of a wide variety of genes via the cAMP-responsible element (CRE) in their promoter. It is the upstream activator CHOP, which encodes a member of the CCAAT/enhancer-binding protein (C/EBP) homologous proteins [61]. This signaling pathway is transcriptionally activated by various cellular stress signals including acute phase response [62]. Physiological dose of DEX treatment was found to abolish the CHOP induction in rats eliciting acute phase response [63]. It appears DEX exerts the same function in TM and turns down this pathway to alleviate the acute phase response gene expression.
SCG2 was down-regulated by 1.56 fold. SCG2 is the precursor of secretoneurin, a neuroendocrine marker expressed in ciliary epithelium [64]. Up-regulated in TM under elevated IOP [23], it likely contributes to coordinate local neurogenic inflammatory responses including vasodilation, increasing vascular permeability and cellular infiltration [65,66]. Thus, DEX suppression of SCG2 might help relieve some inflammation symptoms.
MT1L was up-regulated by 1.90 fold. It is a member of the metallothioneins (MTs) family implicated in disparate physiological functions including zinc and copper metabolism, protection against reactive oxygen species and adaptation to stress [67]. An acute phase response protein, it is induced by inflammatory cytokines [68]. Interestingly, DEX increased MT synthesis via glucocorticoid response elements (GRE) while at the same time suppressed inflammation-induced elevation in MT [69]. It appears DEX exerts opposing effects on MT induction [70]. In the TM, MT has been found up-regulated by elevated IOP23 and DEX treatment [8], which is consistent with our observation. It would be interesting to know which types of MTs are responsible for mediating these two situations and the exact function of MT in TM under DEX treatment or elevated IOP.
HSPA5 was down-regulated by 1.73 fold under DEX treatment. It belongs to the heat shock protein 70 (HSP70) family and resides in the endoplasmic reticulum. HSP70 is induced by a number of environmental and physiological stress stimuli, including inflammation [71]. This down-regulation is probably another mechanism to control the inflammatory response. DEX inhibits major heat shock proteins. Such repression was primarily a glucocorticoid receptor (GR) mediated inhibition of transcription enhancement activity of heat shock transcription factor 1 (HSF1), the upstream transcriptional regulators of HSPs [72].
ASNS/TS11 and WARS were down-regulated by 2.07/2.37 and 1.77 fold, respectively. They are enzymes involved in protein biosynthesis. Their down-regulation in TM by DEX treatment was also observed in the microarray study by Ishibashi et al. [24]. DEX suppression of protein biosynthesis agrees with its anti-inflammatory function through interfering de novo protein biosynthesis [73].
IGFBP2 was up-regulated by 1.70 fold by DEX treatment. The DEX induction of IGFBP2 was also observed in bone marrow stromal cell [74] and TM [24]. IGFBPs comprise a family of insulin-like growth factor (IGF) binding proteins that form complexes with IGFs and moderate their actions in cell [75]. IGFs are involved in the regulation of inflammatory responses as well as immune responses [76]. IGFBPs might exert their anti-inflammatory effects by sequestering the IGFs released during inflammation. IGFBP2 also contains integrin recognition sequence RGD like GAS1. The integrin binding does not require IGF-1. Therefore IGFBPs might also stabilize cell-cell adhesion and decrease the outflow facility. Besides integrin, IGFBP2 also binds to heparin and proteoglycans [77,78]. It is believed that the binding of ECMs and other IGFBP turnover mechanisms can regulate cellular responses by modulating the IGF-1 bioavailability in the pericellular space in vivo. It would be interesting to decipher whether the induction of IGFBP2 by DEX is to counteract or potentiate IGFs' function and whether IGFBP2's binding to integrin can stabilize cell-cell adhesion.
LDHA was upregulated by 1.61 fold. LDH is a tetrameric enzyme with five isoforms composed of combinations of two subunits, LDHA and LDHB. The LDHA subunit carries out anaerobic glycolysis that converts pyruvate to lactate while LDHB converts lactate to pyruate [79]. There are also evidences that LDH plays a regulatory role during gene transcription and DNA replication besides glucose metabolism [80]. In the LDHA promoter there is a silencer module, which can repress cAMP-responsive element (CRE) dependent LDHA expression [81]. The down-regulation of ATF4 (CREB2) shown in our study suggests ATF4 may be responsible for the transcriptional repression of LDHA. The down-regulation of ATF4 would in turn relieve the suppression on LDHA expression. This agrees with the general anti-inflammatory effect of DEX such as reducing protein synthesis, increasing amino acid turnover, and gluconeogenesis. Amino acids catabolism produces pyruvate, which is converted to lactate by LDHA. Lactate is finally transported to liver for gluconeogenesis.
A holistic view of these differentially expressed genes and their biological effects by DEX induction is shown in Figure 4. Except for the MT family protein, all differentially expressed genes have not been previously reported to be regulated by DEX in TM. SCG2, however, was up-regulated in TM under elevated IOP. Obviously a considerable number of DEX regulated genes have overlapping roles in anti-inflammatory response and outflow resistance. The increased outflow resistance and subsequent ocular hypertension may be byproducts of the favorable anti-inflammatory response triggered by DEX treatment. Comparison of this study with an earlier microarray report on elevated IOP in perfused human anterior segment organ culture [23] provides a better understanding on TM responses to different physiological conditions. In the Gonzalez study [23], the stress imposed by elevated IOP on TM triggered the expression of inflammatory genes like interleukin-6, substance P and SCG2, ECM turnover genes like cathepsin-L and stromelysin-1, and stress response genes like metallothionein and αB-crystallin. It appears that TM responses to the pressure by expressing genes to increase the outflow. For example SCG2 was up-regulated in their study while it was down-regulated here. This is because the ultimate product, SN, coordinates inflammatory responses, which are exactly the reactions glucocorticoid is supposed to counteract. Cathepsin-L, a cysteine protease, was up-regulated in the Gonzalez study, while its inhibitor CST3 was up-regulated under DEX treatment in this current study. This CST3 up-regulation might help to increase the outflow in response to the elevated IOP as in the Gonzalez study. The purpose of up-regulating CST3 is likely to inhibit cysteine proteases to reduce inflammatory cellular infiltration. Unfortunately this would increase outflow resistance at the same time. Metallothionein was found to be up-regulated in both studies. However the Gonzalez study did not identify the specific MTs being up-regulated and, at the same time, there appears a complex regulation of MTs' function during inflammation and DEX treatment. Further investigation is necessary to understand MT functions in TM.
It is anomalous that not all people receiving topical glucocorticoid eye drops suffer from steroid-induced ocular hypertension or steroid induced glaucoma. A living system is extremely dynamic and flexible. Tolerance to glucocorticoid is different among people. One must be cautious when extrapolating experimental findings from animal or in vitro study to human or even from a defined group of individuals to the general population.
In contrast to our use of an established steroid responding human cell line that was used for cloning the MYOC gene, Ishibashi et al., established primary TM cell cultures from cadaver eyes and performed gene expression analysis by cDNA microarray [24]. They used four normal human cadaver eyes without any information about their steroid responsiveness. However two of their eye samples expressed much more MYOC than the other two under the same DEX treatment, indicating a possible mixture of two different response groups in their study. Besides, there was no replication for each sample. Although it is understandable that with limited tissue obtainable, replication of a microarray experiment is difficult, yet the statistical power conferred by replication is obvious [25]. Hence, some of their findings can be false positive, as demonstrated by a large standard error of the means. Nonetheless, ours and their study share a few common findings. For example, MYOC and IGFBP2 were found to be up-regulated while ASNS, SCG2, and WARS down-regulated by DEX. The results from these two studies clearly indicate that these genes may participate in the common response pathway to steroid treatment. Therefore we believe both studies have opened up novel research opportunities for glaucoma research.
This current study on cultured human TM cells has demonstrated the power of parallel study on the expression of thousands of genes by microarray technology. Although the number of genes on our array was not particularly high, interesting regulatory mechanisms with rich implications in glaucoma drug development have been revealed. Extending the current study to different TM, ciliary body and ONH [82] cells from various forms of glaucoma and normal people, and to TM cells under different therapeutic drug treatments on higher density microarrays, will reveal further information about the underlying biological pathways. Our understanding on glaucoma pathogenesis will thus be enhanced.
Acknowledgements
This work is funded in part by the WK Lee Eye Foundation, a direct grant from the Medicine Panel and a block grant from the Chinese University of Hong Kong. The authors thank Dr. Thai Nguyen and Dr. Leon Kapp for providing an established human TM cell line and immense support on TM cell culture. We also thank Dr. Ingrid Lönnstedt and Prof Terry Speed for their invaluable comments and technical assistance on using the SMA package, Dr. Bart van der Burg for his comments on the possible regulations of TSC22. Many thanks to Mr. Abel C.S. Chun for his support in tedious literature review. Special thanks to Ms. Alice Y.M. Lee for her assistance in proofreading this manuscript.
References
1. Adcock IM, Caramori G. Cross-talk between pro-inflammatory
transcription factors and glucocorticoids. Immunol Cell Biol 2001;
79:376-84. ![]()
2. Goulding NJ, Euzger HS, Butt SK, Perretti M. Novel pathways for
glucocorticoid effects on neutrophils in chronic inflammation. Inflamm
Res 1998; 47:S158-65. ![]()
3. Armaly MF. Effect of corticosteroids on intra ocular and fluid dynamics. I. The effect of dexamethasone in the normal eye. Arch Ophthalmol 1963; 70:482-91.
4. Shields MB. Steroid-induced glaucoma. In: Shields MB, editors. Textbook of glaucoma. 4th ed. Baltimore: Williams & Willkins; 1998. p. 323-8.
5. Wordinger RJ, Clark AF. Effects of glucocorticoids on the
trabecular meshwork: towards a better understanding of glaucoma. Prog
Retin Eye Res 1999; 18:629-67. ![]()
6. Polansky JR, Fauss DJ, Chen P, Chen H, Lutjen-Drecoll E, Johnson
D, Kurtz RM, Ma ZD, Bloom E, Nguyen TD. Cellular pharmacology and
molecular biology of the trabecular meshwork inducible glucocorticoid
response gene product. Ophthalmologica 1997; 211:126-39.
![]()
7. Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden
SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R,
Kalenak JW, Craven ER, Sheffield VC. Identification of a gene that
causes primary open angle glaucoma. Science 1997; 275:668-70.
![]()
8. Nguyen TD, Chen P, Huang WD, Chen H, Johnson D, Polansky JR. Gene
structure and properties of TIGR, an olfactomedin-related glycoprotein
cloned from glucocorticoid-induced trabecular meshwork cells. J Biol
Chem 1998; 273:6341-50. ![]()
9. Polansky JR, Fauss DJ, Zimmerman CC. Regulation of TIGR/MYOC gene
expression in human trabecular meshwork cells. Eye 2000; 14:503-14.
![]()
10. Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D,
Durcan FJ, McCartney PJ, Mackey DA, Sheffield VC, Stone EM. Clinical
features associated with mutations in the chromosome 1 open-angle
glaucoma gene (GLC1A). N Engl J Med 1998; 338:1022-7. ![]()
11. Lam DS, Leung YF, Chua JK, Baum L, Fan DS, Choy KW, Pang CP.
Truncations in the TIGR gene in individuals with and without primary
open-angle glaucoma. Invest Ophthalmol Vis Sci 2000; 41:1386-91.
![]()
12. Pang CP, Leung YF, Fan B, Baum L, Tong WC, Lee WS, Chua JK, Fan
DS, Liu Y, Lam DS. TIGR/MYOC gene sequence alterations in individuals
with and without primary open-angle glaucoma. Invest Ophthalmol Vis Sci
2002; 43:3231-5. ![]()
13. Sarfarazi M, Child A, Stoilova D, Brice G, Desai T, Trifan OC,
Poinoosawmy D, Crick RP. Localization of the fourth locus (GLC1E) for
adult-onset primary open-angle glaucoma to the 10p15-p14 region. Am J
Hum Genet 1998; 62:641-52. ![]()
14. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M,
Heon E, Krupin T, Ritch R, Kreutzer D, Crick RP, Sarfarazi M.
Adult-onset primary open-angle glaucoma caused by mutations in
optineurin. Science 2002; 295:1077-9. ![]()
15. Raymond V. Molecular genetics of the glaucomas: mapping of the
first five "GLC" loci. Am J Hum Genet 1997; 60:272-7. ![]()
16. Trifan OC, Traboulsi EI, Stoilova D, Alozie I, Nguyen R, Raja S,
Sarfarazi M. A third locus (GLC1D) for adult-onset primary open-angle
glaucoma maps to the 8q23 region. Am J Ophthalmol 1998; 126:17-28.
![]()
17. Wirtz MK, Samples JR, Rust K, Lie J, Nordling L, Schilling K,
Acott TS, Kramer PL. GLC1F, a new primary open-angle glaucoma locus,
maps to 7q35-q36. Arch Ophthalmol 1999; 117:237-41. ![]()
18. Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring
of gene expression patterns with a complementary DNA microarray. Science
1995; 270:467-70. ![]()
19. Duggan DJ, Bittner M, Chen Y, Meltzer P, Trent JM. Expression
profiling using cDNA microarrays. Nat Genet 1999; 21(1 Suppl):10-4.
![]()
20. Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP,
Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES.
Molecular classification of cancer: class discovery and class prediction
by gene expression monitoring. Science 1999; 286:531-7.
![]()
21. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A,
Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore
T, Hudson J Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC,
Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W,
Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of
diffuse large B-cell lymphoma identified by gene expression profiling.
Nature 2000; 403:503-11. ![]()
22. Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett
HA, He YD, Dai H, Walker WL, Hughes TR, Tyers M, Boone C, Friend SH.
Signaling and circuitry of multiple MAPK pathways revealed by a matrix
of global gene expression profiles. Science 2000; 287:873-80.
![]()
23. Gonzalez P, Epstein DL, Borras T. Genes upregulated in the human
trabecular meshwork in response to elevated intraocular pressure. Invest
Ophthalmol Vis Sci 2000; 41:352-61. ![]()
24. Ishibashi T, Takagi Y, Mori K, Naruse S, Nishino H, Yue BY,
Kinoshita S. cDNA microarray analysis of gene expression changes induced
by dexamethasone in cultured human trabecular meshwork cells. Invest
Ophthalmol Vis Sci 2002; 43:3691-7. ![]()
25. Lee ML, Kuo FC, Whitmore GA, Sklar J. Importance of replication
in microarray gene expression studies: statistical methods and evidence
from repetitive cDNA hybridizations. Proc Natl Acad Sci U S A 2000;
97:9834-9. ![]()
26. Lonnstedt I, Speed TP. Replicated Microarray Data. Statistica Sinica 2002; 12:31-46.
27. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Press; 1989.
28. Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP.
Normalization for cDNA microarray data: a robust composite method
addressing single and multiple slide systematic variation. Nucleic Acids
Res 2002; 30:e15. ![]()
29. Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel
RNA-binding protein in human myxoid liposarcoma. Nature 1993; 363:640-4.
![]()
30. Leung YF, Pang CP. EYE on bioinformatics: dissecting complex disease traits in silico. Applied Bioinformatics 2002; 1:69-80.
31. Tomiuk S, Hofmann K. Microarray probe selection strategies. Brief
Bioinform 2001; 2:329-40. ![]()
32. Filla MS, Liu X, Nguyen TD, Polansky JR, Brandt CR, Kaufman PL,
Peters DM. In vitro localization of TIGR/MYOC in trabecular meshwork
extracellular matrix and binding to fibronectin. Invest Ophthalmol Vis
Sci 2002; 43:151-61. ![]()
33. Tawara A, Okada Y, Kubota T, Suzuki Y, Taniguchi F, Shirato S,
Nguyen TD, Ohnishi Y. Immunohistochemical localization of MYOC/TIGR
protein in the trabecular tissue of normal and glaucomatous eyes. Curr
Eye Res 2000; 21:934-43. ![]()
34. Caballero M, Borras T. Inefficient processing of an
olfactomedin-deficient myocilin mutant: potential physiological
relevance to glaucoma. Biochem Biophys Res Commun 2001; 282:662-70.
![]()
35. O'Brien ET, Ren X, Wang Y. Localization of myocilin to the golgi
apparatus in Schlemm's canal cells. Invest Ophthalmol Vis Sci 2000;
41:3842-9. ![]()
36. Mertts M, Garfield S, Tanemoto K, Tomarev SI. Identification of
the region in the N-terminal domain responsible for the cytoplasmic
localization of Myoc/Tigr and its association with microtubules. Lab
Invest 1999; 79:1237-45. ![]()
37. Wentz-Hunter K, Ueda J, Shimizu N, Yue BY. Myocilin is associated
with mitochondria in human trabecular meshwork cells. J Cell Physiol
2002; 190:46-53. ![]()
38. Cai S, Liu X, Glasser A, Volberg T, Filla M, Geiger B, Polansky
JR, Kaufman PL. Effect of latrunculin-A on morphology and
actin-associated adhesions of cultured human trabecular meshwork cells.
Mol Vis 2000; 6:132-43 <http://www.molvis.org/molvis/v6/a18/>. ![]()
39. Del Sal G, Ruaro EM, Utrera R, Cole CN, Levine AJ, Schneider C.
Gas1-induced growth suppression requires a transactivation-independent
p53 function. Mol Cell Biol 1995; 15:7152-60. ![]()
40. Evdokiou A, Cowled PA. Growth-regulatory activity of the growth
arrest-specific gene, GAS1, in NIH3T3 fibroblasts. Exp Cell Res 1998;
240:359-67. ![]()
41. Inuzuka H, Miyatani S, Takeichi M. R-cadherin: a novel
Ca(2+)-dependent cell-cell adhesion molecule expressed in the retina.
Neuron 1991; 7:69-79. ![]()
42. Lecanda F, Cheng SL, Shin CS, Davidson MK, Warlow P, Avioli LV,
Civitelli R. Differential regulation of cadherins by dexamethasone in
human osteoblastic cells. J Cell Biochem 2000; 77:499-506.
![]()
43. Rosenberg P, Esni F, Sjodin A, Larue L, Carlsson L, Gullberg D,
Takeichi M, Kemler R, Semb H. A potential role of R-cadherin in striated
muscle formation. Dev Biol 1997; 187:55-70. ![]()
44. Peterson JA, Tian B, Bershadsky AD, Volberg T, Gangnon RE,
Spector I, Geiger B, Kaufman PL. Latrunculin-A increases outflow
facility in the monkey. Invest Ophthalmol Vis Sci 1999; 40:931-41.
![]()
45. Ueda J, Wentz-Hunter K, Yue BY. Distribution of myocilin and
extracellular matrix components in the juxtacanalicular tissue of human
eyes. Invest Ophthalmol Vis Sci 2002; 43:1068-76. ![]()
46. Alvarado JA, Yun AJ, Murphy CG. Juxtacanalicular tissue in
primary open angle glaucoma and in nonglaucomatous normals. Arch
Ophthalmol 1986; 104:1517-28. ![]()
47. Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M, Grierson I.
Ultrastructure of the trabecular meshwork in untreated cases of primary
open-angle glaucoma (POAG). Exp Eye Res 1993; 56:683-92.
![]()
48. Zhou L, Li Y, Yue BY. Glucocorticoid effects on extracellular
matrix proteins and integrins in bovine trabecular meshwork cells in
relation to glaucoma. Int J Mol Med 1998; 1:339-46. ![]()
49. Steely HT, Browder SL, Julian MB, Miggans ST, Wilson KL, Clark
AF. The effects of dexamethasone on fibronectin expression in cultured
human trabecular meshwork cells. Invest Ophthalmol Vis Sci 1992;
33:2242-50. ![]()
50. Yun AJ, Murphy CG, Polansky JR, Newsome DA, Alvarado JA. Proteins
secreted by human trabecular cells. Glucocorticoid and other effects.
Invest Ophthalmol Vis Sci 1989; 30:2012-22. ![]()
51. Gerber A, Wille A, Welte T, Ansorge S, Buhling F. Interleukin-6
and transforming growth factor-beta 1 control expression of cathepsins B
and L in human lung epithelial cells. J Interferon Cytokine Res 2001;
21:11-9. ![]()
52. Schulte W, Scholze H. Action of the major protease from Entamoeba
histolytica on proteins of the extracellular matrix. J Protozool 1989;
36:538-43. ![]()
53. Sloane BF. Cathepsin B and cystatins: evidence for a role in
cancer progression. Semin Cancer Biol 1990; 1:137-52. ![]()
54. Bjarnadottir M, Grubb A, Olafsson I. Promoter-mediated,
dexamethasone-induced increase in cystatin C production by HeLa cells.
Scand J Clin Lab Invest 1995; 55:617-23. ![]()
55. Clark AF, Wilson K, McCartney MD, Miggans ST, Kunkle M, Howe W.
Glucocorticoid-induced formation of cross-linked actin networks in
cultured human trabecular meshwork cells. Invest Ophthalmol Vis Sci
1994; 35:281-94. ![]()
56. O'Brien ET, Perkins SL, Roberts BC, Epstein DL. Dexamethasone
inhibits trabecular cell retraction. Exp Eye Res 1996; 62:675-88.
![]()
57. Shibanuma M, Kuroki T, Nose K. Isolation of a gene encoding a
putative leucine zipper structure that is induced by transforming growth
factor beta 1 and other growth factors. J Biol Chem 1992; 267:10219-24.
![]()
58. Kester HA, Blanchetot C, den Hertog J, van der Saag PT, van der
Burg B. Transforming growth factor-beta-stimulated clone-22 is a member
of a family of leucine zipper proteins that can homo- and heterodimerize
and has transcriptional repressor activity. J Biol Chem 1999;
274:27439-47. ![]()
59. Ohta S, Shimekake Y, Nagata K. Molecular cloning and
characterization of a transcription factor for the C-type natriuretic
peptide gene promoter. Eur J Biochem 1996; 242:460-6. ![]()
60. Takashima Y, Taniguchi T, Yoshida M, Haque MS, Igaki T, Itoh H,
Nakao K, Honda Y, Yoshimura N. Ocular hypotension induced by
intravitreally injected C-type natriuretic peptide. Exp Eye Res 1998;
66:89-96. ![]()
61. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D.
Regulated translation initiation controls stress-induced gene expression
in mammalian cells. Mol Cell 2000; 6:1099-108. ![]()
62. Sylvester SL, ap Rhys CM, Luethy-Martindale JD, Holbrook NJ.
Induction of GADD153, a CCAAT/enhancer-binding protein (C/EBP)-related
gene, during the acute phase response in rats. Evidence for the
involvement of C/EBPs in regulating its expression [published erratum
appears in: J Biol Chem 1995; 270:14842]. J Biol Chem 1994;
269:20119-25. ![]()
63. Eastman HB, Fawcett TW, Udelsman R, Holbrook NJ. Effects of
perturbations of the hypothalamic-pituitary-adrenal axis on the acute
phase response: altered C/EBP and acute phase response gene expression
in lipopolysaccharide-treated rats. Shock 1996; 6:286-92.
![]()
64. Ortego J, Escribano J, Crabb J, Coca-Prados M. Identification of
a neuropeptide and neuropeptide-processing enzymes in aqueous humor
confers neuroendocrine features to the human ocular ciliary epithelium.
J Neurochem 1996; 66:787-96. ![]()
65. Wiedermann CJ, Dunzendorfer S, Kahler CM, Reinisch N,
Schratzberger P. Secretoneurin and neurogenic inflammation. Zhongguo Yao
Li Xue Bao 1999; 20:789-94. ![]()
66. Collins JJ, Usip S, McCarson KE, Papka RE. Sensory nerves and
neuropeptides in uterine cervical ripening. Peptides 2002; 23:167-83.
![]()
67. Hidalgo J, Aschner M, Zatta P, Vasak M. Roles of the
metallothionein family of proteins in the central nervous system. Brain
Res Bull 2001; 55:133-45. ![]()
68. Min KS, Terano Y, Onosaka S, Tanaka K. Induction of hepatic
metallothionein by nonmetallic compounds associated with acute-phase
response in inflammation. Toxicol Appl Pharmacol 1991; 111:152-62.
![]()
69. Min KS, Mukai S, Ohta M, Onosaka S, Tanaka K. Glucocorticoid
inhibition of inflammation-induced metallothionein synthesis in mouse
liver. Toxicol Appl Pharmacol 1992; 113:293-8. ![]()
70. Min KS, Kim H, Fujii M, Tetsuchikawahara N, Onosaka S.
Glucocorticoids suppress the inflammation-mediated tolerance to acute
toxicity of cadmium in mice. Toxicol Appl Pharmacol 2002; 178:1-7.
![]()
71. Multhoff G, Botzler C. Heat-shock proteins and the immune
response. Ann N Y Acad Sci 1998; 851:86-93. ![]()
72. Wadekar SA, Li D, Periyasamy S, Sanchez ER. Inhibition of heat
shock transcription factor by GR. Mol Endocrinol 2001; 15:1396-410.
![]()
73. Goppelt-Struebe M. Molecular mechanisms involved in the
regulation of prostaglandin biosynthesis by glucocorticoids. Biochem
Pharmacol 1997; 53:1389-95. ![]()
74. Milne M, Quail JM, Rosen CJ, Baran DT. Insulin-like growth factor
binding proteins in femoral and vertebral bone marrow stromal cells:
expression and regulation by thyroid hormone and dexamethasone. J Cell
Biochem 2001; 81:229-40. ![]()
75. Kostecka Y, Blahovec J. Insulin-like growth factor binding
proteins and their functions (minireview). Endocr Regul 1999; 33:90-4.
![]()
76. Heemskerk VH, Daemen MA, Buurman WA. Insulin-like growth factor-1
(IGF-1) and growth hormone (GH) in immunity and inflammation. Cytokine
Growth Factor Rev 1999; 10:5-14. ![]()
77. Arai T, Busby W Jr, Clemmons DR. Binding of insulin-like growth
factor (IGF) I or II to IGF-binding protein-2 enables it to bind to
heparin and extracellular matrix. Endocrinology 1996; 137:4571-5.
![]()
78. Russo VC, Bach LA, Fosang AJ, Baker NL, Werther GA. Insulin-like
growth factor binding protein-2 binds to cell surface proteoglycans in
the rat brain olfactory bulb. Endocrinology 1997; 138:4858-67.
![]()
79. Jungmann RA, Huang D, Tian D. Regulation of LDH-A gene expression
by transcriptional and posttranscriptional signal transduction
mechanisms. J Exp Zool 1998; 282:188-95. ![]()
80. Zhong XH, Howard BD. Phosphotyrosine-containing lactate
dehydrogenase is restricted to the nuclei of PC12 pheochromocytoma
cells. Mol Cell Biol 1990; 10:770-6. ![]()
81. Chung KC, Huang D, Chen Y, Short S, Short ML, Zhang Z, Jungmann
RA. Identification of a silencer module which selectively represses
cyclic AMP-responsive element-dependent gene expression. Mol Cell Biol
1995; 15:6139-49. ![]()
82. Hernandez MR, Agapova OA, Yang P, Salvador-Silva M, Ricard CS,
Aoi S. Differential gene expression in astrocytes from human normal and
glaucomatous optic nerve head analyzed by cDNA microarray. Glia 2002;
38:45-64. ![]()