Received 23 January 2002 | Accepted 20 March 2002 | Published 22 March 2002
Reprint
Received 23 January 2002 | Accepted 20 March 2002 | Published 22 March 2002 |
Download Reprint |
Factors influencing a-crystallin association with phospholipid vesicles
Brian A. Cobb,
J. Mark Petrash
Departments of Ophthalmology & Visual Sciences and Genetics, Washington University School of Medicine, St. Louis, MO
Correspondence to: J. Mark Petrash, Department of Ophthalmology & Visual Sciences, Washington University School of Medicine, 660 South Euclid Avenue, Box 8096, St. Louis, MO, 63110; Phone: (314) 362-1172; FAX: (314) 362-3638; email: petrash@vision.wustl.edu
Abstract
Purpose: Lens lipids undergo a number of changes with age, including an overall increase in phospholipid acyl chain saturation and a decrease in length. In addition, the amount of membrane bound a-crystallin increases dramatically with age and with the onset of cataract. The aim of this study was to determine if a link exists between age and cataract associated changes in lens lipids and the changes in a-crystallin membrane association.
Methods: Protein-free lipid vesicles composed of a wide variety of synthetic and lens-derived lipid vesicles were formed by sonication. These vesicles were used with fluorescent native and recombinant a-crystallin conjugates in vesicle binding assays. Vesicles were collected by centrifugation and bound a-crystallin was quantified with fluorescence intensity measurements.
Results: a-Crystallin complexes showed remarkably similar binding profiles for all lipid vesicles tested, regardless of lipid origin, phospholipid head group, acyl chain length or saturation, and inclusion of cholesterol. In addition, recombinant a-crystallin complexes bind to these vesicles in a manner that is essentially indistinguishable from that of native human and bovine a-crystallins. Unlike a-crystallin binding to lens membranes containing intrinsic proteins, binding of a-crystallin to protein-free vesicles was very high capacity and unsaturable.
Conclusions: We conclude from these data that the binding of a-crystallin to lens membranes is not lipid-specific. Furthermore, protein post-translational changes, such as phosphorylation, do not appear to alter a-crystallin binding to these vesicles. Given the linearity of the binding curves, we propose that the only limiting factor for normal a-crystallin membrane binding is available surface area on the bilayer. Finally, the present data suggests that increased in vivo membrane association of a-crystallin is not a result of lipid changes, but more likely a result of non-lipid factors such as the accumulation of high molecular weight forms of a-crystallin.
Introduction
a-Crystallin is the major protein of vertebrate lenses. It is found in the water soluble and insoluble fractions of lens fiber cells and can represent up to 50% of the total soluble protein [1,2]. a-Crystallin is a large heteromeric complex containing 30 to 40 copies of two closely related subunits, aA- (aA) and aB-crystallin (aB), in roughly a 3:1 ratio in humans [2]. The aA protein is found exclusively in the lens whereas aB is also found in skeletal muscle, skin, brain, and several other tissues [3,4]. Both subunits are members of the small heat shock protein family and can suppress stress-induced protein aggregation in vitro while conferring heat shock tolerance to mammalian tissue culture cells [5-7].
The biological function of a-crystallin in the lens is still not completely evident. Its ability to inhibit stress-induced protein aggregation likely represents a prominent role in the maintenance of lens transparency through blocking the formation of protein inclusion bodies, which could scatter light and cause opacification [5]. However, this chaperone-like activity (CLA) is not the only functional capability observed for a-crystallin. It has been shown that a-crystallin can associate with both lens plasma membranes as well as certain types of intermediate filaments, particularly the beaded filament, yet the functional significance of these interactions is not well understood [8].
As the lens ages, changes are seen in the distribution of a-crystallin within fiber cells. Soluble a-crystallin is slowly lost to the water insoluble fraction of the lens with age and at the onset of cataract; however, the nature of the water insoluble fraction is not well characterized [1,9-12]. Water insoluble a-crystallin could be disorganized aggregates of protein and/or filament- or membrane-bound protein. Interestingly, other in vivo studies have shown that the amount of membrane associated crystallin protein, particularly a-crystallin, increases with age and with the onset of cataract, supporting the hypothesis that water insoluble a-crystallin may actually be membrane bound [13,14]. In addition, our recent data on the characterization of the aA R116C congenital cataract mutant supports a model where increased membrane binding may be a critical event in the mechanism of some forms of cataract [15].
The membranes in the lens fiber cells are quite unusual with respect to other cell membranes. They are extremely rich in aquaporin 0, a lens-specific water channel which makes up nearly 50% of the total intrinsic membrane protein in the lens [16]. Other major protein components include gap junctions and cell adhesion molecules [17]. Interestingly, vertebrate lenses have a peculiar lipid composition (Table 1). An estimated 43% of all phospholipids in the lens is 4,5-dihydrosphingomyelin (DH-SPH) [18-20]. Prior to 1994, this sphingolipid had never been found in a naturally occurring membrane, and yet it is present at extremely high concentrations in the lens [19]. Sphingomyelin (SPH), phosphatidylcholine (PC), and phosphatidylethanolamine (PE) are also major components of lens plasma membrane, collectively representing nearly 37% of the total phospholipid [20]. Another distinctive characteristic of lens membranes is the extreme cholesterol concentration, which ranges from 30 mol% of total lipid at the outer cortex to nearly 60 mol% in the lens nucleus [21]. It has been proposed that the high levels of cholesterol and sphingolipids contribute directly to light refraction and overall lens transparency [21].
Much like the differences in a-crystallin and protein distribution in mammalian lenses, the lipid content also undergoes age and cataract associated alterations. For instance, PC, PE, SPH, and overall chain saturation have all been shown to increase steadily with age [20,22]. Those observations suggest that the structural order of the membranes of old lenses is higher than young lenses [23]. Although no mechanism is known, some have proposed that these changes could lead to an increased risk for cataract formation [23,24].
One previous report showed that the presence of 40 mol% cholesterol in disteroylphosphatidylcholine vesicles antagonizes a-crystallin association [22]. This led to the hypothesis that cholesterol has a potential role in preventing large amounts of a-crystallin from binding to fiber cell membranes. Based on these and other studies, it is reasonable to propose that there could be a link between the observed increase in membrane association of a-crystallin in old and cataractous lenses and changes in the lipid content of the membrane [22]. In order to investigate that proposed linkage, we measured the binding capacity of both recombinant and native a-crystallin samples to a variety of synthetic and native lipid vesicles. We found that lipid differences in the head groups, acyl chains and origin do not significantly affect the binding capacity of either recombinant or native a-crystallins. Our data suggest that changes in the amount of a-crystallin associated with old and cataractous lens membranes, in vivo, do not result from lipid changes, but are more likely a result of non-lipid factors.
Methods
Over-expression and purification of human recombinant aA- and aB-crystallin
cDNA encoding human aA- and aB-crystallins were cloned into the pET23d expression plasmid (Novagen, Madison, WI) and then expressed and purified from E. coli (strain BL21) cultures essentially as described previously [25]. Briefly, primary separations were performed on a DEAE-Sepharose anion exchange column at pH 8.0 and final fractionations were performed on a Sephacryl S400HR (Amersham Biosciences Corp, Piscataway, NJ) column in phosphate buffered saline, PBS (137 mM NaCl; 2.7 mM KCl; 4.3 mM Na2HPO4; 1.4 mM KH2PO4; pH 7.3). All proteins were determined to be >99% homogeneous as judged by the presence of a single band on a coomassie blue stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (data not shown). Proteins were stored at -80 °C until use.
Purification of human and bovine native a-crystallin
Human lens tissue was obtained from the Mid-America Eye Bank and homogenized in gel filtration running buffer (50 mM Tris-HCl, pH 7.6; 50 mM NaCl; 1 mM EDTA) using a glass dounce homogenizer. The lysate was cleared with centrifugation at 15,000x g for 15 min and then loaded on a Superdex 200 FPLC column (Amersham Biosciences Corp). The final a-crystallin pool was determined to be >90% homogeneous as judged from a coomassie blue stained SDS-PAGE gel (data not shown) and was stored at -80 °C until use.
Bovine eyes were obtained at a local meat packing company. Lenses were decapsulated, and the cortical fiber cells dissected away from the nucleus, using a scalpel. The cells were homogenized in membrane buffer A (50 mM Tris-HCl, pH 7.4; 5 mM EDTA; 10 mM dithiothreitol; 0.2% sodium azide) using a glass dounce homogenizer. Once lysed, the cell debris was pelleted by centrifugation at 40,000x g for 30 min. The soluble lysate was then loaded on a Sephacryl S400HR column to separate a-crystallin from the other lens proteins. The final a-crystallin pool was determined to be >90% homogeneous as judged from a coomassie blue stained SDS-PAGE gel (data not shown) and was stored at -80 °C until use.
Protein conjugation to AlexaFluor350TM dye
Purified recombinant aA and aB as well as purified native a-crystallins (bovine and human) were conjugated to the AlexaFluor350TM fluorescent tag (MW=410) as described by the manufacturer (Molecular Probes, Eugene, OR). Conjugated protein was separated from non-reacted AlexaFluor350TM using a pre-packed desalting column according to the manufacturer's protocol (BioRad Econo-Pac 10DG Column, Hercules, CA). The purified AlexaFluor350TM-conjugated a-crystallin subunits were analyzed using A280/A346 readings in a Varian Cary 1E UV/VIS spectrophotometer. Protein concentration and degree of conjugation were determined with the following equations:
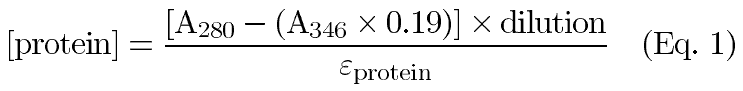
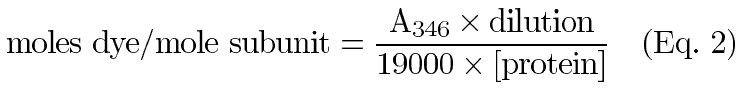
where 0.19 is a correction factor for the absorbance of AlexaFluor350TM at 280 nm, 19000 is the molar extinction coefficient for AlexaFluor350TM, A280 and A346 are the measured absorbance values at 280 and 346 nm respectively, and eprotein is the molar extinction coefficient for a-crystallin. Specific fluorescence activities of the a-crystallin conjugates were determined by analyzing known amounts in a Hoefer Dyna-Quant Spectrofluorometer. The average specific activities were then calculated and expressed in F/nmole protein (F=fluorescence units).
Synthetic lipid vesicle formation
Synthetic lipids were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL) and vesicles were formed using sonication as described by the manufacturer (see Figure 1 for schematic). First, the lipids (in chloroform) were dried down with stirring under an argon stream. Once dried to a film in a glass tube, the lipids were allowed to dry overnight in a lyophilizer to insure complete removal of all organic solvent. Next, the lipid film was rehydrated with stirring in PBS at a temperature greater than the phase transition temperature of the lipid for 1 h. The rehydrated sample was then sonicated using a microtip probe on high for a total of 30 min with a 50% duty cycle (20 s on then 20 s off) to yield small unilamellar vesicles. In most cases, the solution became clear with this treatment. The sample was centrifuged at 10,000x g for 10 min to remove any tip generated debris and nonmembrane lipid aggregates. The cleared solution was aged for at least 24 h before use in the lipid binding experiments described below.
The concentration of each vesicle preparation was quantified by drying aliquots of the vesicle suspension in pre weighed centrifuge tubes in a spin-vacuum (Heto VR1, Denmark). Weight measurements were performed on an analytical balance (Mettler-Toledo, Columbus, OH) and the concentration (nmoles/ml) of membrane in the suspension determined by averaging from at least 4 repetitions.
Native lens lipid extraction and vesicle formation
Membranes were first isolated from bovine cortical fiber cells as previously described by Russell et al [26]. The membrane sample was then extracted with chloroform and methanol as recommended by Bligh and Dyer [27]. First, chloroform and methanol were added to the aqueous membrane suspension to achieve a volumetric ratio of 1:2:0.8 (chloroform:methanol: membranes), which produced a monophasic solution. The sample was then homogenized using a glass dounce homogenizer for 1 min, then diluted with chloroform and dH2O to achieve a 2:2:1.8 volumetric ratio, which forms a biphasic suspension. After the sample was allowed to fully separate, the methanol/dH20 layer was gently removed by aspiration. The chloroform phase was filtered through a DuraporeTM membrane filter (0.22 mm, Millipore, Bedford, MA) and then analyzed using thin layer chromatography developed with chloroform:methanol:ammonium hydroxide at 65:25:4 for the solvent system. Spots were visualized by charring in acid. The dry TLC plate was dipped briefly into 5% H2SO4, then heated on a hot plate for approximately 10 min. Vesicles were made from human lens fiber cell lipids extracted in this manner and analyzed as described above for the synthetic lipid vesicles.
Lipid vesicle binding assays
A varied amount of AlexaFluor350TM conjugated a-crystallins were incubated with lipid vesicles for 15 h at 37 °C to allow binding. Upon completion, each sample was centrifuged for 2 h at 375,000x g to completely pellet the large and small vesicles. The supernatant was decanted and the slightly translucent pellet was resuspended in PBS prior to fluorescence measurement using a Hoefer Dyna-Quant Spectrofluorometer. Solutions containing resuspended pellets were essentially clear. The amount of a-crystallin bound was determined by dividing the total fluorescence intensity of the resuspended vesicles by the specific fluorescence activity of the a-crystallin conjugate. Control binding assays were carried out with AlexaFluor350TM conjugated a-crystallins in the absence of vesicles. Less than 1% of input crystallin was found to sediment in the absence of vesicles under these conditions. Linear regression analysis was used to fit the data sets from varying phospholipid vesicles. Statistical analyses comparing the linear regressions from only 2 plots were performed using a student's T test at a 95% confident level, while comparison of multiple fits (greater than 2) was performed using a nonparametric Kruskal-Wallis test with a 95% confidence level.
Results
Human recombinant a-crystallin association with lipid vesicles
To determine the influence of acyl chain length and saturation as well as lipid head group on the binding properties of recombinant a-crystallin complexes, binding assays were performed using a variety of synthetic vesicles (1,2-dipalmitoyl-SN- glycero-3-phosphatidylcholine, PC 16:0; 1,2-dipalmitelaidoyl-SN-glycero-3-phospha-tidylcholine (trans orientation), PC 16:1t; 1,2-diarachindoyl-SN-glycero-3-phosphatidylcholine, PC 20:0; 1,2-diarachidonoyl-SN-glycero-3-phosphatidylcholine (all cis orientation), PC 20:4c; and SPH) with recombinant AlexaFluor350TM-conjugated aA and aB homocomplexes as well as a reconstituted 3:1 heterocomplex. All binding assays were performed in PBS at pH 7.3 with 5 mM MgCl2 for 15 h at 37 °C. As shown in Figure 2A and Figure 2C, no significant difference was observed for the binding of either aA homocomplexes or 3:1 heterocomplexes to any of the PC vesicles tested (p=0.0937 and 0.7709 respectively). However, a statistically significant difference was seen for these two a-crystallin complexes when binding to PC vesicles was compared to SPH vesicles (p=0.0045 for aA and 0.0162 for 3:1 heterocomplex). For aB, Figure 2B shows that association to PC 16:0, PC 20:4c, and SPH vesicles was indistinguishable (p=0.4492), but differences in binding to PC 16:1t and PC 20:0 were significantly different compared to PC 16:0 (p=0.0142 and 0.0082 respectively). In addition, the binding curves are linear, unlike the binding to native lens membranes [25]. Most data points represent the average of at least 4 assays.
The effect of cholesterol on the association of human recombinant a-crystallin with lipid vesicles
Significant differences between a-crystallin binding to synthetic lipid vesicles with and without 40 mol% cholesterol have been reported [22]. To further probe the effects of cholesterol on a-crystallin binding, recombinant a-crystallin complexes were used in vesicle binding experiments with both PC and SPH vesicles with and without 40 mol% cholesterol. For aA, the presence of cholesterol increases the binding to SPH (p=0.0023) but not PC (p=0.0904) vesicles to a small but statistically significant amount (Figure 3A). aB vesicle binding (Figure 3B) was decreased by the presence of cholesterol in PC but not SPH vesicles (p=0.0001 and 0.0815 respectively), while 3:1 heterocomplex binding (Figure 3C) was not significantly affected by cholesterol in either PC or SPH vesicles (p=0.0713 and 0.1916 respectively). The binding of a-crystallin in all assays remained unsaturable and linear throughout the range assayed.
Native bovine and human a-crystallin vesicle association
To measure the effect of posttranslational modifications commonly found in the lens-derived a-crystallin proteins, native bovine and human a-crystallins were purified by size (approximately 600 to 700 kDa average molecular mass, data not shown), conjugated with AlexaFluor350TM and assayed for PC 16:0 vesicle binding. Figure 4 shows that while native a-crystallin binding is lower than that with recombinant a-crystallins (p=0.0001), proteins derived from both sources produced linear, nonsaturable binding curves over the range of concentrations tested. Further, no difference in vesicle binding was seen between the native human and bovine a-crystallins (p=0.0965).
Lens membrane lipid extraction and vesicle formation
Within the lens, the lipid content is far more complex than the tested synthetic lipid vesicles. In addition, lens plasma membranes contain a high concentration of DH-SPH, which is not commercially available [18,19]. To identify any differences between lens derived lipids and synthetic vesicles, bovine lens membranes were fractionated as described previously, then used as a source of native lens lipids in the extraction procedure described in the Experimental Procedures [26]. Thin layer chromatography (TLC) was used as a means to verify the efficacy of the extraction procedure (Figure 5). The analysis of Rf values obtained demonstrated the presence of key lipids in our sample as compared to the known lipid content of lens cortical fiber cells (Table 2). These lipids were then used to form vesicles for binding experiments.
Native bovine lens lipid vesicle binding
Vesicles composed of native bovine lens lipids were formed and used in binding experiments to measure the effect of lipid vesicle complexity on a-crystallin binding. The binding curves for the native lens lipid vesicles were compared to SPH/Cholesterol vesicles, which are the synthetic vesicles in this study closest to the in vivo lipid composition. Our results for recombinant aA showed no significant difference in binding to either of these vesicle types (p=0.6345, Figure 6A); however, a statistically significant change was seen for both recombinant aB and the reconstituted 3:1 heterocomplex (p<0.0001 for both proteins, Figure 6B and Figure 6C).
Discussion
Previous studies using phospholipid vesicles as a binding template for a-crystallin have been conflicting. Some studies have indicated the requirement of proteins, namely MP26, for binding of a-crystallin to lipid vesicles [28]. Ifeanyi et al. showed significant saturable binding by aA but not aB to vesicles void of protein [29]. Tang et al. demonstrated a strong influence of cholesterol on the binding of a-crystallin that was proposed to be dependent on lipid structural order [22]. In studies where a-crystallin binding to protein free lipid vesicles was observed, saturation was always seen with aA and native a-crystallin and only sometimes with aB [22,29-31]. In the present study, we verified significant binding by both native and recombinant a-crystallin complexes to lipid vesicles void of protein, while observing a general lack of lipid specificity in those interactions.
Lipid order has been previously postulated to be involved with the age dependent increase in the amount of a-crystallin found on lens fiber cell plasma membranes [22]. To address this hypothesis, synthetic vesicles were formed with varying lipid structural order by varying the length and saturation of the acyl chains on commercially available phospholipids. Assays were performed with recombinant a-crystallin complexes (homo- and heterocomplexes) and PC vesicles composed of a variety of acyl chains. We found that changing the acyl chains on the PC head group from dipalmitoyl (PC 16:0) to dipalmitelaidoyl (PC 16:1t), which have phase transition temperatures of 41 °C and -36 °C respectively, does not alter the overall amount of either aA or the 3:1 heterocomplex that is able to associate (Figure 2). Interestingly, aB does show a statistically significant difference in binding to these two vesicles; however, the change is quite small in the context of the amount that associates with lens membranes. Summarized in Table 3, we illustrate the considerable differences in the binding capacities of these vesicles compared to lens derived membranes. Given these differences, it is difficult to imagine that the slight difference in linear-regression slope is biologically meaningful, despite the statistics. Indeed, we found that further alterations to both the acyl chain length, saturation, and even head group also did not dramatically affect the number of binding sites able to accommodate a-crystallin complexes (Figure 2). These data collectively suggest that a-crystallin binding is independent of lipid bilayer fluidity and overall structural order.
Inclusion of cholesterol is also known to affect the lipid structural order of bilayers, particularly in sphingomyelin vesicles (reviewed in [32]). Interestingly, it had also been shown that for some types of vesicles, inclusion of cholesterol antagonizes a-crystallin association [22]. We found that the presence of 40 mol% cholesterol in PC and SPH vesicles, which is similar to the in vivo concentrations within cortical fiber cell membranes, did not dramatically affect the amount of recombinant a-crystallin that was able to bind (Figure 3). Nonetheless, we found that for aA, the presence of cholesterol statistically increases the amount of protein that is able to bind vesicles made of SPH while aB shows a statistically significant decrease in binding. Again, we believe that the small but significant differences seen in these data are negligible in the context of lens membrane binding (Table 3).
From these binding experiments, we conclude that in a synthetic lipid system, lipid order, bilayer fluidity and cholesterol do not play major roles in determining the specificity or amount of a-crystallin association with the membrane. This experimental approach, however, does not control for in vivo posttranslational changes to the protein, such as phosphorylation, nor for the far more complex lipid content of the lens plasma membrane, particularly the inclusion of DH-SPH. To measure any differences that might not be adequately controlled in this recombinant and synthetic system, native a-crystallin and native lens lipids were purified and used in further binding experiments. First, native human and bovine a-crystallin were used in binding experiments with synthetic PC 16:0 vesicles. As before, we found some statistically significant changes in the plots, but no major differences were seen between the amount of native protein able to bind these vesicles as compared to the recombinant a-crystallins tested (Figure 4). This suggests that posttranslational modifications in native sized a-crystallin complexes play little or no role in modulating membrane association. Second, native lens lipid vesicles were used in binding experiments with recombinant a-crystallin complexes (Figure 6). For aA, no significant change between native bovine lipid and synthetic SPH/Cholesterol vesicle binding were seen. Both aB and the reconstituted 3:1 heterocomplex showed significant but not dramatic differences in binding. Based on these data, we conclude that the precise lipid content of target membranes, particularly the head group distribution, is largely irrelevant for a-crystallin association.
We have previously shown that recombinant a-crystallin associates with lens membranes in a saturable manner [15,25]. The present data indicate that the binding to protein free lipid vesicles exceeds 100 fold more than the binding seen with extracted lens membranes containing the usual assortment of lens intrinsic membrane proteins (Table 3). It is this very large difference that leads us to propose that the relatively small but statistically significant differences seen between the binding plots of the various lipid vesicles presented here are biologically irrelevant. Nonetheless, we cannot completely rule out that lipid content does not play a minor role in membrane binding of a-crystallin, however any effect it may have would more likely be indirect (e.g. lipid influences on the packing of protein domains on the membrane).
In vivo studies have shown that the amount of a-crystallin found on the membranes of lens fiber cells dramatically increases with both age and the onset of cataract [13,14]. It has been postulated that changes in the membrane order via changes in the lipid content, including cholesterol, could be a part of the mechanism of increased a-crystallin binding and ultimately cataract pathogenesis [22]. Due to the lack of major differences seen in a-crystallin binding to a variety of vesicles, including ones made of native lens lipids, our results do not support previous hypotheses on a correlation between changes in lipid content, increased a-crystallin membrane association, and cataract. Interestingly, the lack of major differences between recombinant and native a-crystallin in our studies further suggest that posttranslational modifications that do not alter the size of the complex, particularly phosphorylation, do not dramatically alter binding to the membrane. In light of our findings, it appears that the mechanism of change seen in the amount of a-crystallin associated with fiber cell membranes in old and cataractous lenses is more likely due to damage associated protein changes in the crystallin population rather than in the lipids. Indeed, we have recently been able to show that events related to the chaperone like activity and the formation of high molecular weight a-crystallin complexes can explain at least some of the increased membrane association seen in old and cataractous lenses [33]. It is also important to note that our data does not rule out the possibility that lipids may affect lens transparency through direct changes in the refractive properties of lens membranes [24].
We previously demonstrated that trypsinization increased the membrane binding capacity for recombinant a-crystallin, which led to the conclusion that intrinsic membrane proteins hinder the approach of a-crystallin to the lipid bilayer [25]. With past and current data demonstrating significant binding of a-crystallin to protein free synthetic vesicles, it is clear that a-crystallin is directly interacting with the lipid bilayer as opposed to membrane proteins as previously suggested [28]. In addition, the linearity of these data implies that the amount of a-crystallin able to bind to lipid vesicles and lens membranes is only limited by the surface area of lipid bilayer available to a-crystallin. Therefore, we conclude that the interaction is fundamentally nonspecific in nature. This explains why trypsinization of lens membranes, which would likely result in reduced steric hindrance for the approach of a-crystallin to the membrane surface, increases the binding capacity of the recombinant a-crystallin heterocomplex [25]. In addition, lens membranes are more than 50% intrinsic protein by weight, which would lead to very limited amounts of protein free lipid domains within the bilayer, thus accounting for the small binding capacity observed with lens membranes as compared to the protein free vesicles used in this study (see Table 3) [34].
Differences between our observations and those made by previous investigators may be related in part to differences in both the methods used to prepare vesicles and in the conditions for collecting vesicles during the binding assays. Our method utilized probe sonication of a lipid suspension which usually results in vesicles with diameters ranging from 15 to 50 nm. In contrast, previous investigators used an extrusion method for vesicle preparation [22,30]. Although extrusion results in large vesicles with diameters typically in excess of 250 nm, aging small sonicated vesicles (at least 24 h at 4 °C in the present study) increases the mean diameter of these vesicles rapidly, particularly at temperatures below the lipid phase transition temperature (Avanti Polar Lipids, Catalog VI, p. 167-76). It is possible that the vesicles used here and those used in previous reports may show different binding properties due to size variability, however, it is difficult to propose how the potential size difference could account for the significant difference in binding itself. If vesicle size impacted the binding of a-crystallin, the binding measured from day to day would not be consistent with a given vesicle sample stored at 4 °C. Our data remained reproducible for at least one week following vesicle formation, strongly suggesting that within this broad size range, vesicle diameter does not significantly effect binding. In addition, we found that extended centrifugation at high gravitational force was necessary to reproducibly pellet all liposomes present in our lipid suspensions. Although 375,000x g for 2 h is much faster and longer than previously used, we found that these spins introduced less than 1% error in the data due to protein pelleting (data not shown). Given the relatively milder centrifugation conditions used in previous studies, it seems possible that the entire amount of bound a-crystallin could have been underestimated due to incomplete pelleting of the vesicles. When compared to previous studies, our results suggest that far more a-crystallin can bind to protein free lipid vesicles than previously appreciated [28-31].
Despite our previous conclusion that saturable binding to lens membranes implies specificity [25], the present study suggests that a-crystallin membrane association is largely independent of lipid content and is limited only by the available lipid bilayer surface area. As a direct extension of that conclusion, we believe that intrinsic membrane proteins reduce the binding capacity of a-crystallin under normal conditions through simple steric hindrance to the lipids themselves. Therefore, we propose that a-crystallin membrane binding is a nonspecific interaction. Finally, the present data strongly supports our recent report that suggests the increase in a-crystallin membrane association in old and/or cataractous lenses is due to changes in the lens protein population, particularly the formation of chaperone mediated high molecular weight complexes.
Acknowledgements
We wish to thank Dr. Steven Bassnett and Lori Kreisman for a critical reading of this manuscript and Elizabeth Kelley for technical assistance. We also would like to thank Greg Tochtrop and Dr. Douglas Covey for providing expertise and equipment for TLC analysis of the native lens lipids. Finally, we thank Drs. Douglas Borchman and Richard Cenedella for helpful discussions on this manuscript. This work was supported in part by National Institutes of Health Grants EY50673, EY02687, EY06901, DK20579, and an award to the Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness, Inc.
References
1. Bessems GJ, De Man BM, Bours J, Hoenders HJ. Age-related
variations in the distribution of crystallins within the bovine lens.
Exp Eye Res 1986; 43:1019-30. ![]()
2. de Jong WW. Evolution of lens and crystallins. In: Bloemendal H, editor. Molecular and Cellular Biology of the Eye Lens. New York: John Wiley & Sons, Inc.; 1981. p. 221-78.
3. Bhat SP, Nagineni CN. alpha B subunit of lens-specific protein
alpha- crystallin is present in other ocular and non-ocular tissues.
Biochem Biophys Res Commun 1989; 158:319-25. ![]()
4. Bhat SP, Horwitz J, Srinivasan A, Ding L. Alpha B-crystallin
exists as an independent protein in the heart and in the lens. Eur J
Biochem 1991; 202:775-81. ![]()
5. Horwitz J. Alpha-crystallin can function as a molecular chaperone.
Proc Natl Acad Sci U S A 1992; 89:10449-53. ![]()
6. Blackburn R, Galoforo S, Berns CM, Ireland M, Cho JM, Corry PM,
Lee YJ. Thermal response in murine L929 cells lacking alpha B-crystallin
expression and alpha B-crystallin expressing L929 transfectants. Mol
Cell Biochem 1996; 155:51-60. ![]()
7. van den IJssel PR, Overkamp P, Knauf U, Gaestel M, de Jong WW.
Alpha A- crystallin confers cellular thermoresistance. FEBS Lett 1994;
355:54-6. ![]()
8. FitzGerald PG, Graham D. Ultrastructural localization of alpha A-
crystallin to the bovine lens fiber cell cytoskeleton. Curr Eye Res
1991; 10:417-36. ![]()
9. Babizhayev MA, Bours J, Utikal KJ. Isoelectric focusing of
crystallins in microsections of calf and adult bovine lens.
Identification of water-insoluble crystallins complexing under
nondenaturing conditions: demonstration of chaperone activity of
alpha-crystallin. Ophthalmic Res 1996; 28365-74. ![]()
10. Bessems GJ, Hoenders HJ, Wollensak J. Variation in proportion and
molecular weight of native crystallins from single human lenses upon
aging and formation of nuclear cataract. Exp Eye Res 1983; 37:627-37.
![]()
11. Bindels JG, Bours J, Hoenders HJ. Age-dependent variations in the
distribution of rat lens water-soluble crystallins. Size fractionation
and molecular weight determination. Mech Ageing Dev 1983; 21:1-13.
![]()
12. Bours J, Fodisch HJ, Hockwin O. Age-related changes in water and
crystallin content of the fetal and adult human lens, demonstrated by a
microsectioning technique. Ophthalmic Res 1987; 19:235-9.
![]()
13. Boyle DL, Takemoto L. EM immunolocalization of alpha-crystallins:
association with the plasma membrane from normal and cataractous human
lenses. Curr Eye Res 1996; 15:577-82. ![]()
14. Cenedella RJ, Fleschner CR. Selective association of crystallins
with lens 'native' membrane during dynamic cataractogenesis. Curr Eye
Res 1992; 11:801-15. ![]()
15. Cobb BA, Petrash JM. Structural and functional changes in the
alpha A- crystallin R116C mutant in hereditary cataracts. Biochemistry
2000; 39:15791-8. ![]()
16. Keeling P, Johnson K, Sas D, Klukas K, Donahue P, Johnson R.
Arrangement of MP26 in lens junctional membranes: analysis with
proteases and antibodies. J Membr Biol 1983; 74:217-28.
![]()
17. Goodenough DA. The crystalline lens. A system networked by gap
junctional intercellular communication. Semin Cell Biol 1992; 3:49-58.
![]()
18. Ferguson SR, Borchman D, Yappert MC. Confirmation of the identity
of the major phospholipid in human lens membranes. Invest Ophthalmol Vis
Sci 1996; 37:1703-6. ![]()
19. Byrdwell WC, Borchman D, Porter RA, Taylor KG, Yappert MC.
Separation and characterization of the unknown phospholipid in human
lens membranes. Invest Ophthalmol Vis Sci 1994; 35:4333-43.
![]()
20. Merchant TE, Lass JH, Meneses P, Greiner JV, Glonek T. Human
crystalline lens phospholipid analysis with age. Invest Ophthalmol Vis
Sci 1991; 32:549-55. ![]()
21. Borchman D, Cenedella RJ, Lamba OP. Role of cholesterol in the
structural order of lens membrane lipids. Exp Eye Res 1996; 62:191-7.
![]()
22. Tang D, Borchman D, Yappert MC, Cenedella RJ. Influence of
cholesterol on the interaction of alpha-crystallin with phospholipids.
Exp Eye Res 1998; 66:559-67. ![]()
23. Borchman D, Ozaki Y, Lamba OP, Byrdwell WC, Yappert MC. Age and regional structural characterization of clear human lens lipid membranes by infrared and near-infrared Raman spectroscopes. Biospectroscopy 1996; 2:113-23.
24. Paterson CA, Zeng J, Husseini Z, Borchman D, Delamere NA, Garland
D, Jimenez-Asensio J. Calcium ATPase activity and membrane structure in
clear and cataractous human lenses. Curr Eye Res 1997; 16:333-8.
![]()
25. Cobb BA, Petrash JM. Characterization of alpha-crystallin-plasma
membrane binding. J Biol Chem 2000; 275:6664-72. ![]()
26. Russell P, Robison WG Jr, Kinoshita JH. A new method for rapid
isolation of the intrinsic membrane proteins from lens. Exp Eye Res
1981; 32:511-6. ![]()
27. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Canadian Journal of Biochemistry and Physiology 1959; 37:911-7.
28. Mulders JW, Stokkermans J, Leunissen JA, Benedetti EL, Bloemendal
H, de Jong WW. Interaction of alpha-crystallin with lens plasma
membranes. Affinity for MP26. Eur J Biochem 1985; 152:721-8.
![]()
29. Ifeanyi F, Takemoto L. Specificity of alpha crystallin binding to
the lens membrane. Curr Eye Res 1990; 9:259-65. ![]()
30. Borchman D, Tang D. Binding capacity of alpha-crystallin to
bovine lens lipids. Exp Eye Res 1996; 63:407-10. ![]()
31. Ifeanyi F, Takemoto L. Interaction of lens crystallins with lipid
vesicles. Exp Eye Res 1991; 52:535-8. ![]()
32. Slotte JP. Sphingomyelin-cholesterol interactions in biological
and model membranes. Chem Phys Lipids 1999; 102:13-27.
![]()
33. Cobb BA, Petrash JM. alpha-Crystallin chaperone-like activity and
membrane binding in age-related cataracts. Biochemistry 2002; 41:483-90.
![]()
34. Bloemendal H, Zweers A, Vermorken F, Dunia I, Benedetti EL. The
plasma membranes of eye lens fibres. Biochemical and structural
characterization. Cell Differ 1972; 1:91-106. ![]()