Received 29 June 2005 | Accepted 13 December 2005 | Published 19 December 2005
Reprint
Received 29 June 2005 | Accepted 13 December 2005 | Published 19 December 2005 |
Download Reprint |
Disease-associated mutations in CNGB3 produce gain of function alterations in cone cyclic nucleotide-gated channels
Scott R. Bright,1,2 Travis E. Brown,1,2
Michael D. Varnum1,2,3
1Department of Veterinary and Comparative Anatomy, Pharmacology and Physiology, 2Program in Neuroscience, and 3Center for Integrative Biotechnology, Washington State University, Pullman, WA
Correspondence to: Michael D. Varnum, Washington State University, PO Box 646520, Pullman, WA, 99164; Phone: (509) 335-0701; FAX: (509) 335-4650; email: varnum@wsu.edu
Abstract
Purpose: To characterize the functional consequences of disease-associated mutations in the CNGB3 (B3) subunit of human cone photoreceptor cyclic nucleotide-gated channels in order to gain insight into disease mechanisms.
Methods: Three separate disease-associated mutations were generated in CNGB3: F525N, R403Q, and T383fsX. These mutant subunits were then heterologously expressed in Xenopus oocytes in combination with wild type CNGA3 (A3) subunits, and characterized by patch-clamp recording in the inside-out configuration.
Results: Co-expression of A3 and B3F525N, A3 and B3R403Q, or A3 and B3R403Q and B3T383fsX subunits resulted in channels that exhibited an increase in ligand sensitivity without a reduction in current density compared to wild-type heteromeric channels. Each simulated disease state produced channels that exhibited greater sensitivity to block by L-cis-diltiazem than homomeric CNGA3 channels, confirming that the mutant CNGB3 subunits were competent to form functional heteromeric channels. Each combination of subunits displayed an increase in apparent affinity for cGMP relative to wild-type heteromeric channels. However, F525N enhanced cGMP apparent affinity to a significantly greater extent than the other two modeled disease states.
Conclusions: We have examined the gating effects of two previously uncharacterized disease-associated mutations in the CNGB3 subunit and found that in each case, the mutations resulted in a gain of function molecular phenotype. Furthermore, the magnitude of the effect on channel function correlated with the severity of the associated disease. The complete achromatopsia-associated F525N mutation resulted in more pronounced alterations in channel function than the mutation combinations linked to macular degeneration or progressive cone dystrophy.
Introduction
Visual impairment is one of the most common causes of disability, and inherited forms of retinal diseases afflict approximately 100,000 people in the United States, according to the NEI National Plan for Eye and Vision Research (2004). A subset of these inherited disorders directly impact the function and survival of cone photoreceptors, or of cone and rod photoreceptors together, leading to impairment or loss of color and high-acuity vision. These include a range of progressive and stationary cone dystrophies or dysfunctions, such as macular degeneration, progressive cone dystrophy, and achromatopsia [1,2]. Of the diseases that alter cone function, complete achromatopsia is one of the most severe, characterized by a total absence of cone function apparent from birth, nystagmus and photophobia [3].
Genetic studies of patients with achromatopsia have identified three important loci. Specifically, mutations in the CNGA3 and CNGB3 genes encoding the subunits of cone cyclic nucleotide-gated (CNG) channels [4-11], and GNAT2, the gene encoding the α subunit of cone-specific transducin [12,13], have been linked to this disease. Mutations in the genes encoding the cone CNG channel subunits are particularly prevalent in patients with achromatopsia, as two separate studies have found that 25-40% of these patients have mutations in CNGA3 and 40-50% have mutations in CNGB3 [10,11]. In addition, recent studies examining the consequences of a CNGA3 knockout in mice found that these mice exhibited a phenotype that resembled complete achromatopsia in humans [14,15]. Furthermore, these mice exhibited alterations beyond the function of CNG channels, such that the targeting of cone opsins, the expression of other visual cascade proteins, and the morphological development of cone somata were also perturbed [14,15]. These results highlight the importance of CNG channels for normal visual transduction and for the pathophysiology of retinal disease.
CNG channels are a critical component of visual transduction involved in converting light-induced changes in intracellular cGMP concentration into electrical responses that can be later interpreted by the brain as visual information. CNGA3 subunits can form functional homomeric channels when heterologously expressed alone, while CNGB3 subunits cannot. Heteromeric cone CNG channels are thought to be composed of two CNGA3 subunits and two CNGB3 subunits [16]; the co-expression of CNGB3 subunits with CNGA3 results in channels exhibiting properties that more closely resemble those of native channels, including enhanced cAMP efficacy and sensitivity to L-cis-diltiazem block [17,18].
Each CNG channel subunit is thought to contain six transmembrane regions (S1-S6) with a pore-forming (P) re-entrant loop between S5 and S6. Additionally, these subunits have intracellular amino and carboxy termini that are critical to CNG channel gating, with the carboxy-terminal region of each subunit containing a cyclic nucleotide binding domain (CNBD) [19]. The binding of cGMP or cAMP to the CNBD is coupled to an allosteric transition that results in channel opening [20]. The gating of CNG channels is also slightly sensitive to voltage, a phenomenon that results in outward rectification of CNG channel currents [21-23]. CNG channels are nonselective cation channels permeable to Na+, K+, and Ca2+ ions. Calcium entering through CNG channels can modulate phototransduction sensitivity via multiple mechanisms, including the binding of calcium-calmodulin to channels to down regulate their activity [18,24-26].
In addition to their association with achromatopsia, mutations in CNGB3 have also been linked to progressive cone dystrophy and macular degeneration [27,28]. We hypothesize that the severity of the functional alterations in the mutant channels parallels the severity of the associated retinal diseases. This hypothesis has not yet been addressed as relatively few disease-associated mutations in CNGB3 have been functionally characterized and the three mutations that have been studied to date are associated with complete achromatopsia [29,30]. In this study, we address this hypothesis by examining two previously uncharacterized CNGB3 mutations, F525N and R403Q, which are associated with complete achromatopsia and macular degeneration, respectively, in patients homozygous for these mutant alleles [10,28]. In addition, we have characterized channels that model a compound heterozygous genotype associated with progressive cone dystrophy [27] by co-expressing CNGB3R403Q and CNGB3T383fsX subunits with wild-type CNGA3. These studies help to verify the pathogenic nature of the F525N and R403Q mutations and provide clues about the resulting pathogenic mechanisms.
Methods
Molecular biology
A cDNA clone for human CNGB3 (AF272900) was isolated as previously described (18). Human CNGA3 (AF065314) [31,32] was a generous gift of Prof. K.-W. Yau (John Hopkins University, Baltimore, MD). Both CNGB3 and CNGA3 were subcloned into pGEMHE [33] for heterologous expression in Xenopus oocytes. Xenopus oocytes were isolated as previously described [34]. The animal use protocols were consistent with the recommendations of the American Veterinary Medical Association and were approved by the IACUC of Washington State University (Pullman, WA). Point mutations were generated by overlapping polymerase chain reaction (PCR) [35]. For all mutations, the amplified cassettes were sequenced to confirm the fidelity of the PCR. For expression in Xenopus oocytes, mRNA was synthesized in vitro from channel subunit cDNA using an upstream T 7 promoter and the mMessage mMachine kit (Ambion, Austin, TX). RNA was injected into each oocyte at approximately 8 ng of CNGA3 and 20 ng of total CNGB3 [36]. This ratio has been previously shown to efficiently generate heteromeric channels [16].
Electrophysiology
Electrophysiological recordings were made two to seven days after injection. Patch-clamp experiments were performed with an Axopatch 200 B patch-clamp amplifier (Axon Instruments, Foster City, CA) in the inside-out configuration. Initial pipette resistances were 0.45-0.75 Mohm. Currents were low-pass filtered at 2 kHz and sampled at 25 kHz. Intracellular and extracellular solutions contained 130 mM NaCl, 0.2 mM EDTA, and 3 mM HEPES (pH 7.2). Cyclic nucleotides (Sigma, St. Louis, MO) were added to intracellular solutions as indicated. L-cis-diltiazem (BIOMOL, Plymouth Meeting, PA) was applied at a concentration of 25 μM in the presence of 1 mM cGMP to confirm the formation of heteromeric channels. Intracellular solutions were changed using an RSC-160 rapid solution changer (Molecular Kinetics, Pullman, WA). Currents in the absence of cyclic nucleotide were subtracted. Recordings were made at 20 to 22 °C. Dose-response relationships were obtained by plotting the steady-state current at +80 mV as a function of cyclic nucleotide concentration. Dose-response data were fitted to the Hill equation:
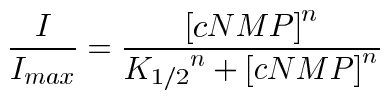
where I is the current amplitude, Imax is the maximum current, [cNMP] is the ligand concentration, K1/2 is the apparent affinity for ligand, and n is the Hill coefficient. Data were acquired using Pulse (HEKA Elektronik, Lambrecht, Germany), and analyzed using Igor Pro (Wavemetrics, Lake Oswego, OR) and SigmaPlot (SPSS, Chicago, IL). The data were expressed as mean±SEM unless otherwise indicated. Statistical significance was determined using a Student's t-test or a Mann-Whitney rank sum test (SigmaStat, SPSS), and a p value less than 0.05 was considered significant.
Results
We determined the functional consequences of disease-associated mutations in the CNGB3 subunit by co-expressing mutant subunits with wild-type CNGA3 in Xenopus oocytes at a constant ratio of CNGA3 to total CNGB3. Specifically, we examined channels containing CNGB3F525N [10], a mutation located in the C-linker region near the cyclic nucleotide-binding domain, CNGB3R403Q [27,28], a mutation located in the alpha-helical portion of the P-loop, and CNGB3T383fsX [10,11,27], a previously characterized null mutation [29] that results in truncation of the entire carboxy-terminal cytoplasmic domain plus S6 and the P-loop (Figure 1). There are several ways in which channel mutations can result in cone dysfunction, including disruption of heteromeric channel assembly, alterations in protein stability and expression levels, or, alternatively, mutant subunits might form functional channels with altered properties.
To help distinguish among these possibilities, we first determined if the mutant CNGB3 subunits could be incorporated into functional channels by using the CNG channel blocker L-cis-diltiazem as a reporter for heteromeric channel formation (Figure 2A,B). This compound, when applied to the cytoplasmic face of the membrane, blocks native and recombinant heteromeric CNG channels in a voltage-dependent manner. Heterologously expressed CNGA3 homomeric channels exhibit little sensitivity to L-cis-diltiazem, while wild type heteromeric channels are significantly inhibited [18]. We found that channels formed by CNGB3-F525N or CNGB3-R403Q subunits in combination with CNGA3 subunits exhibited significantly greater diltiazem inhibition relative to homomeric CNGA3 channels (Figure 2A,B), indicating that the mutant CNGB3 subunits participated in the formation of functional heteromeric channels. However, mutant channels exhibited somewhat reduced inhibition relative to wild type heteromeric channels.
Next we tested the possibility that these mutations altered the stability or plasma membrane expression level of the mutant subunits by determining the impact of the mutations on normalized patch-current density in a saturating concentration of cGMP (Figure 2C). Only the F525N mutation altered this parameter, resulting in a significant enhancement of expression level relative to wild type heteromeric channels. None of the mutations or mutant subunit combinations reduced patch-current density. These results indicated that neither protein expression level nor apparent stability were significantly reduced by these mutations, suggesting that they instead exert their putative pathogenic effects by altering the gating or ion permeation properties of the channels. A possible caveat related to this finding is that channel expression levels in Xenopus oocytes may or may not replicate expression levels in specialized photoreceptor cells.
Wild type heteromeric cone CNG channels typically exhibit outward rectification in their steady-state currents in saturating cGMP (I+80/I-80=1.36±0.029; N=13) that is thought to result from the slight voltage sensitivity of CNG channel gating [21-23]. We found that the mutations characterized in this study significantly altered the rectification properties of the resulting channels in opposing directions. Compared to wild-type heteromeric channels, F525N-containing channels displayed significantly reduced outward rectification (I+80/I-80=1.21±0.023, N=7 and p=0.003), while R403Q-containing channels exhibited significantly increased rectification (I+80/I-80=1.99±0.086, N=16 and p<0.001 for the homozygous model and I+80/I-80=1.84±0.149, N=8 and p=0.005 for the compound heterozygous model, as illustrated in Figure 2A and Figure 3A). These alterations in channel rectification may reflect either changes in the ion permeation pathway or secondary effects of changes in channel gating.
We next examined the gating properties of the mutant channels by studying their activation by ligand. A large change in the intrinsic gating properties of the channels would be expected to alter the efficacy of the partial agonist cAMP relative to the nearly full agonist cGMP. Again, only the F525N mutation significantly altered this parameter compared to wild type heteromeric channels (p<0.001; Figure 3). However, all mutant groups exhibited significantly higher relative cAMP efficacy compared to homomeric channels, providing additional evidence for heteromeric channel formation with these mutant CNGB3 subunits.
To further characterize changes in gating of the mutant channels, we examined the apparent affinity for both cGMP and cAMP. Apparent affinity is influenced by both initial ligand binding and the subsequent allosteric transition(s) associated with channel opening. Hence, if these mutations reduce the energy barrier for channel opening, they would be expected to increase the apparent affinity for ligand. Each mutant construct significantly enhanced the apparent affinity for cGMP (Figure 4A,C; R403Q: K1/2=13.8±0.6 μM, n=2.0±0.1; R403Q+T383fsX: K1/2=13.7±0.7 μM, n=2.0±0.1; F525N: K1/2=6.2±0.6 μM, n=1.5±0.1) relative to wild type heteromeric channels (K1/2=18.4±1.0 μM, n=1.9±0.1). Furthermore, the apparent affinity for cGMP was significantly greater for the F525N group relative to the other mutant groups, and this mutation significantly altered the Hill coefficient (n) for the cGMP dose response curve relative to wild-type channels (p=0.003). In addition to these changes in cGMP apparent affinity, the F525N mutation also significantly enhanced the apparent affinity for cAMP relative to wild-type channels (Figure 4B,D). These results collectively indicate that the mutations in CNGB3 resulted in gain-of-function alterations in channel gating that appear to correlate with the severity of the associated disease. The gating effects for these mutations and other disease-associated CNGB3 mutations that have been characterized previously are summarized in Table 1.
Discussion
We have examined changes in channel gating for recombinant cone CNG channels having disease-associated mutations in the CNGB3 subunit: F525N, R403Q, or a combination of R403Q plus T383fsX subunits. These CNGB3 mutations have been linked to inherited retinal diseases of complete achromatopsia, macular degeneration, and progressive cone dystrophy, respectively [10,27,28]. Complete achromatopsia represents the most severe retinal disorder in this group as it is characterized by a complete loss of cone function [3], whereas patients having CNGB3 mutations associated with cone dystrophy or macular degeneration exhibit evidence of residual cone function, including greater visual acuity and color discrimination compared to patients with achromatopsia [28,37].
We have found that the CNGB3 mutations examined in this study all produce gain-of-function changes in channel gating. Furthermore, none of the simulated disease states decreased functional expression level and the F525N mutation increased current density. These results are consistent with enhanced channel activity. All disease-associated mutations in CNGB3 that have been functionally characterized to date [29,30], including the mutations examined here, produce gain-of-function effects on channel gating (Table 1). Even effectively null CNGB3 mutations, including T383fsX and another frameshift mutation that results in a severely truncated CNGB3 subunit [38], may cause gain-of-function changes in channel gating, since homomeric CNGA3 channels exhibit increased cGMP apparent affinity compared to heteromeric channels and cannot be down regulated by calcium-calmodulin [18,29,39]. Null mutations, however, in CNGB3 may disturb the stability and/or localization of CNGA3 subunits in the photoreceptor outer segment as has been demonstrated recently for CNGA1 subunits in rods of CNGB1-/- mice [2].
Typically, gain-of-function mutations in ion channels are associated with autosomal dominant or semi-dominant patterns of inheritance [40], but the diseases associated with these CNGB3 mutations appear to follow an autosomal recessive inheritance pattern [10,11,28]. There is limited evidence, however, of haploinsufficiency associated with the T383fsX mutation, as a recent report described two individuals with only one mutant allele (T383fsX/+) that exhibited mild reductions in visual acuity [28]. However, the possibility remains that the phenotype in these individuals is the result of an additional, unidentified mutation in some other gene or in a critical noncoding region of CNGB3, as other individuals heterozygous for this allele were asymptomatic [28].
Another key finding in the present study is that the severity of the retinal disorder caused by the mutation or combination of mutations correlated well with the severity of the functional consequences of the mutation(s) for channel gating (Table 1). Thus, the mutation associated with complete achromatopsia (F525N) had a greater impact on apparent affinity for cGMP than the other two disease-related, mutant subunit combinations. In addition, F525N was the only mutation in this study that significantly altered sensitivity to cAMP. Enhanced cAMP sensitivity may potentiate responses to cGMP at physiological ligand concentrations [41-43]. These findings, in combination with previously published results characterizing other CNGB3 mutations [29,30], suggest that the degree of pathogenesis resulting from the CNGB3 mutations arises primarily from the functional alterations in the channel itself. Additional functional studies for other disease associated mutations in the CNGB3 subunit are needed to verify this observation. This relationship between disease severity and channel gating perturbation may not apply to disorders linked to mutations in CNGA3 [9,44].
While we have observed a correlation between disease severity and the functional alterations in mutant channels, we did not observe any additional effects of co-expression of CNGB3T383fsX with CNGB3R403Q and CNGA3WT compared to the CNGB3R403Q+CNGA3WT channels. Patients that are compound heterozygotes for both mutant alleles exhibit relatively more severe deficits in cone function compared to patients homozygous for the R403Q allele [27,28]. One possible explanation for this discrepancy is that the severity of the disease in compound heterozygotes is influenced by an unidentified pathogenic or modifying mutation elsewhere. Another possibility is that the T383fsX allele, which is thought to essentially represent a null mutation [29], influences disease progression via a gene-dosage effect. A single R403Q allele may produce insufficient quantities of functional CNGB3, a haplo-insufficiency. While we did not observe any additional consequences of CNGB3T383fsX co-expression on current density, our heterologous expression system may inadequately model the expression pattern present in cone photoreceptors of compound heterozygotes.
While the crystal structures of eukaryotic CNG channel subunits have not been solved, insight into the mechanisms by which these mutations alter channel function is provided by the known structures of related channels (Figure 1). For example, the crystal structure of the carboxy-terminal region of the hyperpolarization-gated, cyclic nucleotide modulated channel, HCN2, represents a structural model of the same region of CNG channels [45], including the C-linker where F525 is located. The gating of CNG channels has been previously shown to be highly sensitive to structural alterations in the C-linker [46-55]. The HCN2 crystal structure is thought to represent the ligand-bound but closed state of the channel [45,53,54]. Disruption of C-linker salt bridges evident in the HCN2 crystal structure enhances open probability in both CNGA1 and HCN2 channels, suggesting that these charge pairs stabilize the closed conformation of the C-linker [53]. F525 is predicted to participate in the high degree of packing observed in this region; conversion of this highly conserved hydrophobic residue into a polar asparagine might disrupt packing and thereby destabilize the closed conformation of the channel.
The crystal structures of related potassium channels, KcsA and MthK [56-58], provide some insight into the mechanism of R403Q's functional effects. By analogy to these structures, R403 appears to be located in the α-helical region of the P-loop. Although the P-loop is generally considered to be part of the "pore-forming" region of the channel, the α-helical portion does not directly line the pore. Furthermore, the pattern of cysteine accessibility to methanethiosulfonate (MTS) reagents in this region of CNG channels is consistent with an α-helical structure and suggests that R403 (L356 in CNGA1) is not accessible to the ion conduction pathway [59-61]. The pattern of cysteine accessibility was also dependent on the conformational state of the channel, which suggests that this region undergoes a structural transition during channel opening [59,60]. Furthermore, the pore helix has been shown to be an important site for inter-subunit contacts within the tetrameric channel [56]. Hence, the R403Q mutation could impact channel gating by altering the nature of the interaction between CNG channel subunits.
The possibility remains that R403Q could modify the channel's ion conduction properties indirectly by changing the structure of the pore region or by changing the electrostatic environment surrounding the critical pore glutamate residues in such a manner that the calcium binding and permeation properties of the CNG channels are altered [62]. Also, the electrostatic properties and orientation of the pore helix have been shown to influence the "cation-attractive" nature of the pore region in the potassium channel KcsA [56,63]. Additional experiments are needed to determine if R403Q alters permeation properties such as calcium block, ion selectivity, and single-channel conductance. Of possible relevance to this question, R403Q alters the rectification of the CNG channel currents in a surprising manner. A gain-of-function gating effect could reduce outward rectification, as did the F525N mutation, by making the allosteric transition associated with channel opening more favorable. In contrast to this, we found that R403Q increased outward rectification. R403Q might indirectly alter the structure of an ion binding site in the channel pore.
The changes in apparent affinity for ligand discussed above may reflect changes in the initial docking of ligand, the subsequent conformational change(s) associated with channel opening, or both [64]. For the F525N mutation, increased cAMP efficacy along with increased apparent affinity for cGMP provide evidence that the intrinsic gating properties of the channels are indeed altered. This conclusion is consistent with the location of F525N (and R403Q) outside of the CNBD. Single-channel recordings are needed to verify this interpretation.
Gain of function mutations in CNGB3 are expected to result in enhanced channel activity in cone photoreceptors. Since the channels are steeply sensitive to cGMP and physiological ligand concentrations are well below the channels' K1/2 for cGMP, even small changes in ligand sensitivity may have profound cellular consequences. Similarly, mutations in proteins that regulate photoreceptor cGMP levels, such as those that produce constitutive guanylyl cyclase activity [65,66] or loss of cGMP phosphodiesterase activity [67], may result in inappropriate levels of channel activity. As CNG channels represent the pathway for calcium entry into the photoreceptor outer segment, an increase in the number of open channels may produce elevated intracellular calcium levels and subsequently, cone dysfunction or degeneration. Sustained elevation of intracellular calcium has been suggested to be a crucial step for apoptosis in general [68], and has been linked specifically to rod photoreceptor degeneration via apoptosis [69].
In summary, we have characterized gating effects of disease-associated mutations in the CNGB3 subunit and found that they exhibit a gain-of-function phenotype at the molecular level. Furthermore, the mutation associated with the more severe disorder of complete achromatopsia had more dramatic consequences for channel gating compared to those mutations linked to macular degeneration or progressive cone dystrophy. To our knowledge, this study represents the first functional characterization of mutations in CNGB3 that are associated with macular degeneration and progressive cone dystrophy, and thus provides new insight into their molecular pathophysiology. Additional studies are needed to verify the possible mechanisms for cellular pathogenesis proposed here.
Acknowledgements
We are grateful to E. D. Rich for excellent technical assistance, to C. Peng and C. Liu for helpful discussions, and to all three colleagues and to L. K. Sprunger for their comments on the manuscript. We also thank K.-W. Yau for sharing the cDNA clone for human CNGA3. This work was supported by grants from the National Eye Institute (R01EY12836) to M. D. Varnum and from the Poncin Foundation to S. R. Bright.
References
1. Pacione LR, Szego MJ, Ikeda S, Nishina PM, McInnes RR. Progress
toward understanding the genetic and biochemical mechanisms of inherited
photoreceptor degenerations. Annu Rev Neurosci 2003; 26:657-700.
![]()
2. Huttl S, Michalakis S, Seeliger M, Luo DG, Acar N, Geiger H, Hudl
K, Mader R, Haverkamp S, Moser M, Pfeifer A, Gerstner A, Yau KW, Biel M.
Impaired channel targeting and retinal degeneration in mice lacking the
cyclic nucleotide-gated channel subunit CNGB1. J Neurosci 2005;
25:130-8. ![]()
3. Simunovic MP, Moore AT. The cone dystrophies. Eye 1998; 12:553-65.
![]()
4. Kohl S, Marx T, Giddings I, Jagle H, Jacobson SG, Apfelstedt-Sylla
E, Zrenner E, Sharpe LT, Wissinger B. Total colourblindness is caused by
mutations in the gene encoding the alpha-subunit of the cone
photoreceptor cGMP-gated cation channel. Nat Genet 1998; 19:257-9.
![]()
5. Wissinger B, Jagle H, Kohl S, Broghammer M, Baumann B, Hanna DB,
Hedels C, Apfelstedt-Sylla E, Randazzo G, Jacobson SG, Zrenner E, Sharpe
LT. Human rod monochromacy: linkage analysis and mapping of a cone
photoreceptor expressed candidate gene on chromosome 2q11. Genomics
1998; 51:325-31. ![]()
6. Winick JD, Blundell ML, Galke BL, Salam AA, Leal SM, Karayiorgou
M. Homozygosity mapping of the Achromatopsia locus in the Pingelapese.
Am J Hum Genet 1999; 64:1679-85. ![]()
7. Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U,
Spegal R, Anastasi M, Zrenner E, Sharpe LT, Wissinger B. Mutations in
the CNGB3 gene encoding the beta-subunit of the cone photoreceptor
cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to
chromosome 8q21. Hum Mol Genet 2000; 9:2107-16. ![]()
8. Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN, Mitchell TN, Silva ED,
Maumenee IH. Genetic basis of total colourblindness among the
Pingelapese islanders. Nat Genet 2000; 25:289-93. ![]()
9. Wissinger B, Gamer D, Jagle H, Giorda R, Marx T, Mayer S, Tippmann
S, Broghammer M, Jurklies B, Rosenberg T, Jacobson SG, Sener EC,
Tatlipinar S, Hoyng CB, Castellan C, Bitoun P, Andreasson S, Rudolph G,
Kellner U, Lorenz B, Wolff G, Verellen-Dumoulin C, Schwartz M, Cremers
FP, Apfelstedt-Sylla E, Zrenner E, Salati R, Sharpe LT, Kohl S. CNGA3
mutations in hereditary cone photoreceptor disorders. Am J Hum Genet
2001; 69:722-37. ![]()
10. Johnson S, Michaelides M, Aligianis IA, Ainsworth JR, Mollon JD,
Maher ER, Moore AT, Hunt DM. Achromatopsia caused by novel mutations in
both CNGA3 and CNGB3. J Med Genet 2004; 41:e20. ![]()
11. Kohl S, Varsanyi B, Antunes GA, Baumann B, Hoyng CB, Jagle H,
Rosenberg T, Kellner U, Lorenz B, Salati R, Jurklies B, Farkas A,
Andreasson S, Weleber RG, Jacobson SG, Rudolph G, Castellan C, Dollfus
H, Legius E, Anastasi M, Bitoun P, Lev D, Sieving PA, Munier FL, Zrenner
E, Sharpe LT, Cremers FP, Wissinger B. CNGB3 mutations account for 50%
of all cases with autosomal recessive achromatopsia. Eur J Hum Genet
2005; 13:302-8. ![]()
12. Aligianis IA, Forshew T, Johnson S, Michaelides M, Johnson CA,
Trembath RC, Hunt DM, Moore AT, Maher ER. Mapping of a novel locus for
achromatopsia (ACHM4) to 1p and identification of a germline mutation in
the alpha subunit of cone transducin (GNAT2). J Med Genet 2002;
39:656-60. ![]()
13. Kohl S, Baumann B, Rosenberg T, Kellner U, Lorenz B, Vadala M,
Jacobson SG, Wissinger B. Mutations in the cone photoreceptor G-protein
alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet
2002; 71:422-5. ![]()
14. Biel M, Seeliger M, Pfeifer A, Kohler K, Gerstner A, Ludwig A,
Jaissle G, Fauser S, Zrenner E, Hofmann F. Selective loss of cone
function in mice lacking the cyclic nucleotide-gated channel CNG3. Proc
Natl Acad Sci U S A 1999; 96:7553-7. ![]()
15. Michalakis S, Geiger H, Haverkamp S, Hofmann F, Gerstner A, Biel
M. Impaired opsin targeting and cone photoreceptor migration in the
retina of mice lacking the cyclic nucleotide-gated channel CNGA3. Invest
Ophthalmol Vis Sci 2005; 46:1516-24. ![]()
16. Peng C, Rich ED, Varnum MD. Subunit configuration of heteromeric
cone cyclic nucleotide-gated channels. Neuron 2004; 42:401-10.
![]()
17. Gerstner A, Zong X, Hofmann F, Biel M. Molecular cloning and
functional characterization of a new modulatory cyclic nucleotide-gated
channel subunit from mouse retina. J Neurosci 2000; 20:1324-32.
![]()
18. Peng C, Rich ED, Thor CA, Varnum MD. Functionally important
calmodulin-binding sites in both NH2- and COOH-terminal regions of the
cone photoreceptor cyclic nucleotide-gated channel CNGB3 subunit. J Biol
Chem 2003; 278:24617-23. ![]()
19. Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels.
Physiol Rev 2002; 82:769-824. ![]()
20. Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu
Rev Cell Dev Biol 2003; 19:23-44. ![]()
21. Karpen JW, Zimmerman AL, Stryer L, Baylor DA. Gating kinetics of
the cyclic-GMP-activated channel of retinal rods: flash photolysis and
voltage-jump studies. Proc Natl Acad Sci U S A 1988; 85:1287-91.
![]()
22. Zagotta WN, Siegelbaum SA. Structure and function of cyclic
nucleotide-gated channels. Annu Rev Neurosci 1996; 19:235-63.
![]()
23. Benndorf K, Koopmann R, Eismann E, Kaupp UB. Gating by cyclic GMP
and voltage in the alpha subunit of the cyclic GMP-gated channel from
rod photoreceptors. J Gen Physiol 1999; 114:477-90. ![]()
24. Chen TY, Illing M, Molday LL, Hsu YT, Yau KW, Molday RS. Subunit
2 (or beta) of retinal rod cGMP-gated cation channel is a component of
the 240-kDa channel-associated protein and mediates Ca(2+)-calmodulin
modulation. Proc Natl Acad Sci U S A 1994; 91:11757-61.
![]()
25. Hackos DH, Korenbrot JI. Calcium modulation of ligand affinity in
the cyclic GMP-gated ion channels of cone photoreceptors. J Gen Physiol
1997; 110:515-28. ![]()
26. Rebrik TI, Korenbrot JI. In intact mammalian photoreceptors,
Ca2+-dependent modulation of cGMP-gated ion channels is detectable in
cones but not in rods. J Gen Physiol 2004; 123:63-75. ![]()
27. Michaelides M, Aligianis IA, Ainsworth JR, Good P, Mollon JD,
Maher ER, Moore AT, Hunt DM. Progressive cone dystrophy associated with
mutation in CNGB3. Invest Ophthalmol Vis Sci 2004; 45:1975-82.
![]()
28. Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, Dryja TP. Cone
cGMP-gated channel mutations and clinical findings in patients with
achromatopsia, macular degeneration, and other hereditary cone diseases.
Hum Mutat 2005; 25:248-58. ![]()
29. Peng C, Rich ED, Varnum MD. Achromatopsia-associated mutation in
the human cone photoreceptor cyclic nucleotide-gated channel CNGB3
subunit alters the ligand sensitivity and pore properties of heteromeric
channels. J Biol Chem 2003; 278:34533-40. ![]()
30. Okada A, Ueyama H, Toyoda F, Oda S, Ding WG, Tanabe S, Yamade S,
Matsuura H, Ohkubo I, Kani K. Functional role of hCngb3 in regulation of
human cone cng channel: effect of rod monochromacy-associated mutations
in hCNGB3 on channel function. Invest Ophthalmol Vis Sci 2004;
45:2324-32. ![]()
31. Yu WP, Grunwald ME, Yau KW. Molecular cloning, functional
expression and chromosomal localization of a human homolog of the cyclic
nucleotide-gated ion channel of retinal cone photoreceptors. FEBS Lett
1996; 393:211-5. ![]()
32. Wissinger B, Muller F, Weyand I, Schuffenhauer S, Thanos S, Kaupp
UB, Zrenner E. Cloning, chromosomal localization and functional
expression of the gene encoding the alpha-subunit of the cGMP-gated
channel in human cone photoreceptors. Eur J Neurosci 1997; 9:2512-21.
![]()
33. Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian
K+ channel determined by construction of multimeric cDNAs. Neuron 1992;
9:861-71. ![]()
34. Varnum MD, Black KD, Zagotta WN. Molecular mechanism for ligand
discrimination of cyclic nucleotide-gated channels. Neuron 1995;
15:619-25. ![]()
35. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed
mutagenesis by overlap extension using the polymerase chain reaction.
Gene 1989; 77:51-9. ![]()
36. White MM, Mayne KM, Lester HA, Davidson N. Mouse-Torpedo hybrid
acetylcholine receptors: functional homology does not equal sequence
homology. Proc Natl Acad Sci U S A 1985; 82:4852-6. ![]()
37. Sloan LL, Feiock K. Selective impairment of cone function.
Perimetric techniques for its detection. Mod Probl Ophthalmol 1972;
11:50-62. ![]()
38. Rojas CV, Maria LS, Santos JL, Cortes F, Alliende MA. A
frameshift insertion in the cone cyclic nucleotide gated cation channel
causes complete achromatopsia in a consanguineous family from a rural
isolate. Eur J Hum Genet 2002; 10:638-42. ![]()
39. Grunwald ME, Zhong H, Lai J, Yau KW. Molecular determinants of
the modulation of cyclic nucleotide-activated channels by calmodulin.
Proc Natl Acad Sci U S A 1999; 96:13444-9. ![]()
40. Lester HA, Karschin A. Gain of function mutants: ion channels and
G protein-coupled receptors. Annu Rev Neurosci 2000; 23:89-125.
![]()
41. Orr HT, Lowry OH, Cohen AI, Ferrendelli JA. Distribution of
3':5'-cyclic AMP and 3':5'-cyclic GMP in rabbit retina in vivo:
selective effects of dark and light adaptation and ischemia. Proc Natl
Acad Sci U S A 1976; 73:4442-5. ![]()
42. Furman RE, Tanaka JC. Photoreceptor channel activation:
interaction between cAMP and cGMP. Biochemistry 1989; 28:2785-8.
![]()
43. He Y, Karpen JW. Probing the interactions between cAMP and cGMP
in cyclic nucleotide-gated channels using covalently tethered ligands.
Biochemistry 2001; 40:286-95. ![]()
44. Liu C, Varnum MD. Functional consequences of progressive cone
dystrophy-associated mutations in the human cone photoreceptor cyclic
nucleotide-gated channel CNGA3 subunit. Am J Physiol Cell Physiol 2005;
289:C187-98. ![]()
45. Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E.
Structural basis for modulation and agonist specificity of HCN pacemaker
channels. Nature 2003; 425:200-5. ![]()
46. Gordon SE, Zagotta WN. Subunit interactions in coordination of
Ni2+ in cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A 1995;
92:10222-6. ![]()
47. Zong X, Zucker H, Hofmann F, Biel M. Three amino acids in the
C-linker are major determinants of gating in cyclic nucleotide-gated
channels. EMBO J 1998; 17:353-62. ![]()
48. Paoletti P, Young EC, Siegelbaum SA. C-Linker of cyclic
nucleotide-gated channels controls coupling of ligand binding to channel
gating. J Gen Physiol 1999; 113:17-34. ![]()
49. Broillet MC. A single intracellular cysteine residue is
responsible for the activation of the olfactory cyclic nucleotide-gated
channel by NO. J Biol Chem 2000; 275:15135-41. ![]()
50. Johnson JP Jr, Zagotta WN. Rotational movement during cyclic
nucleotide-gated channel opening. Nature 2001; 412:917-21.
![]()
51. Rosenbaum T, Gordon SE. Dissecting intersubunit contacts in
cyclic nucleotide-gated ion channels. Neuron 2002; 33:703-13.
![]()
52. Zhou L, Olivier NB, Yao H, Young EC, Siegelbaum SA. A conserved
tripeptide in CNG and HCN channels regulates ligand gating by
controlling C-terminal oligomerization. Neuron 2004; 44:823-34.
![]()
53. Craven KB, Zagotta WN. Salt bridges and gating in the
COOH-terminal region of HCN2 and CNGA1 channels. J Gen Physiol 2004;
124:663-77. ![]()
54. Hua L, Gordon SE. Functional interactions between A' helices in
the C-linker of open CNG channels. J Gen Physiol 2005; 125:335-44.
![]()
55. Johnson JP Jr, Zagotta WN. The carboxyl-terminal region of cyclic
nucleotide-modulated channels is a gating ring, not a permeation path.
Proc Natl Acad Sci U S A 2005; 102:2742-7. ![]()
56. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen
SL, Chait BT, MacKinnon R. The structure of the potassium channel:
molecular basis of K+ conduction and selectivity. Science 1998;
280:69-77. ![]()
57. Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal
structure and mechanism of a calcium-gated potassium channel. Nature
2002; 417:515-22. ![]()
58. Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open
pore conformation of potassium channels. Nature 2002; 417:523-6.
![]()
59. Liu J, Siegelbaum SA. Change of pore helix conformational state
upon opening of cyclic nucleotide-gated channels. Neuron 2000;
28:899-909. ![]()
60. Giorgetti A, Nair AV, Codega P, Torre V, Carloni P. Structural
basis of gating of CNG channels. FEBS Lett 2005; 579:1968-72.
![]()
61. Becchetti A, Gamel K, Torre V. Cyclic nucleotide-gated channels.
Pore topology studied through the accessibility of reporter cysteines. J
Gen Physiol 1999; 114:377-92. ![]()
62. Seifert R, Eismann E, Ludwig J, Baumann A, Kaupp UB. Molecular
determinants of a Ca2+-binding site in the pore of cyclic
nucleotide-gated channels: S5/S6 segments control affinity of intrapore
glutamates. EMBO J 1999; 18:119-30. ![]()
63. Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+
channel: electrostatic stabilization of monovalent cations. Science
1999; 285:100-2. ![]()
64. Colquhoun D. Binding, gating, affinity and efficacy: the
interpretation of structure-activity relationships for agonists and of
the effects of mutating receptors. Br J Pharmacol 1998; 125:924-47.
![]()
65. Dizhoor AM. Regulation of cGMP synthesis in photoreceptors: role
in signal transduction and congenital diseases of the retina. Cell
Signal 2000; 12:711-9. ![]()
66. Olshevskaya EV, Ennilov AN, Dizhoor AM. Factors that affect
regulation of cGMP synthesis in vertebrate photoreceptors and their
genetic link to human retinal degeneration. Mol Cell Biochem 2002;
230:139-47. ![]()
67. Rattner A, Sun H, Nathans J. Molecular genetics of human retinal
disease. Annu Rev Genet 1999; 33:89-131. ![]()
68. Nicotera P, Orrenius S. The role of calcium in apoptosis. Cell
Calcium 1998; 23:173-80. ![]()
69. Tsang SH, Gouras P, Yamashita CK, Kjeldbye H, Fisher J, Farber
DB, Goff SP. Retinal degeneration in mice lacking the gamma subunit of
the rod cGMP phosphodiesterase. Science 1996; 272:1026-9.
![]()