Received 26 December 1997 | Accepted 22 May 1998 | Published 18 June 1998
Reprint
Received 26 December 1997 | Accepted 22 May 1998 | Published 18 June 1998 |
Download Reprint |
Local microdomain structure in the terminal extensions of ßA3- and ßB2-crystallins
Yuri V. Sergeev,1
Larry L. David,2
Harry C. Chen,3
John N. Hope,4
J. Fielding Hejtmancik1
1National Eye Institute, NIH, Bethesda, MD; 2Departments of Oral Molecular Biology and Ophthalmology, Oregon Health Sciences University, Portland, OR; 3National Institute of Child Health and Development, NIH, Bethesda, MD; 4MedImmune Inc., Gaithersburg, MD
Correspondence to: Yuri V. Sergeev, OGCSB/NEI/NIH, 10/10B10, 9000 Rockville Pike, Bethesda, MD, 20892-1860; email: sergeev@helix.nih.gov
Abstract
Purpose: Although the crystal structures of the core domains of bovine ßB2-crystallin have been determined and those of other ß[gamma]-crystallins modeled, the positions of the N- and C-termini are not resolvable by X-ray crystallography. Here we model the possible structural organization of the terminal arms of mouse ßA3- and ßB2-crystallins and test this model against the results of partial proteolysis.
Methods: The secondary structure of the terminal extensions was predicted by 3 different methods, one a nearest-neighbor method modified to use overlapping sequence tripeptides. Recombinant ßA3- and ßB2-crystallins were expressed using baculovirus vectors in S. frugiperda Sf9 cells. Crystallins were sequenced by the Edman degradation method.
Results: The N-terminal extension of ßB2-crystallin includes a series of hydrophilic residues from Q-11 to Q-9 which have high propensity of a helical conformation. The N-terminal arm of ßA3-crystallin is also predicted to have two helical segments, from Q-24 to E-20 and M-13 to A-12. Partial characterization of the baculovirus extract showed a thiol protease inhibited by leupeptin and E-64. As predicted by the model, recombinant ßB2-crystallin subjected to partial proteolysis was cleaved adjacent to the helical domain, while the N-terminal cleavage site in recombinant ßA3-crystallin was within 1 residue of an interhelical junction. Our model also predicts the products of partial proteolytic degradation of ßB2- and ßA3-crystallins from human, rat, bovine and chicken lenses incubated with the protease m-calpain.
Conclusions: These results suggest the existence of local microdomain structures in the N- and C-terminal extensions of ßA3- and ßB2-crystallins, which appear to be more susceptible to proteolytic degradation in regions adjacent to these putative domains.
Introduction
The ß[gamma]-crystallins of the eye lens form a gene superfamily, sharing a common core structure composed of four Greek key motifs forming two domains. However the ß-crystallins have N- and C-terminal extensions ('arms') while the [gamma]-crystallins have minimal or no extensions. The ß-crystallin family consists of two groups of proteins: acidic ßA1-, ßA2-, ßA3-, ßA4- and basic ßB1-, ßB2-, ßB3-crystallins [1-3]. Acidic ß-crystallins have only N-terminal extensions while basic ß-crystallins have both N- and C-terminal extensions. While the crystallographic structure of ßB2-crystallin has been determined [4], the terminal 8 and 10 residues of the amino- and carboxy-terminal arms respectively can not be resolved on electron density maps, suggesting that they do not occupy a fixed position. NMR study of ßB2-crystallin in solution shows that the terminal extensions possess little ordered structure, are accessible to solvent, and flex freely from the main body of the protein [5]. However the question of the terminal extensions' structure and their possible role in ß-crystallin function are still largely unresolved.
[gamma]-Crystallins, which are present in monomeric form in lens cell extracts, have only rudimentary terminal extensions. The protein surface areas involved in interactions between the domains of the [gamma]-crystallins and domains of different molecules of ß-crystallin dimers show a high degree of the sequence similarity [6]. The similarity of these surface areas explains why ß-crystallins form both homo- and hetero-dimers easily. The presence of terminal arms in oligomeric ß-crystallins and their absence from the monomeric [gamma]-crystallins suggests that the arms may have a role in stabilizing the structure of ß-crystallin oligomers [7,8]. The terminal arms of ß-crystallins appear to be lost as lens fibers age and in some forms of cataract [9]. Depending on the specific amino acid left exposed, cutting terminal arms may lead to inappropriate aggregation and insolubility of the ß-crystallins [10]. The importance of terminal extensions in the lens was emphasized in a study by David et al. [11], who showed that specific cleavage of 4 to 49 residues from ß-crystallin amino-terminal extensions may result from activation of the protease m-calpain and correlates with protein insolubilization and cataract formation. Although the terminal extensions in ß-crystallins appear not to form a single stable structure, the specific cleavage pattern seen with a variety of proteases suggests the presence of structural domains located between protease cleavage sites in the extensions. Here we apply secondary structure prediction methods to find common structural patterns in the terminal extensions and correlate these patterns with the sites at which proteases cleave the terminal extensions.
We used three approaches to locate elements of possible secondary structure in the terminal extensions. First, we applied traditional methods including the PHD secondary structure prediction method [12] and NNSSP, which use a nearest-neighbor algorithm [13]. Both methods give a predictive accuracy close to 70% when using information from multiple sequence alignments. We also used a modification of the nearest-neighbor algorithm in which the length of the test segment was reduced to 3 peptides to allow consideration of only local interactions between residues in the arms. In addition, when the sequences of the ßB2-crystallin amino- and carboxy-terminal arms were aligned inversely, correlating with their positions in the structure of the ß-crystallin dimer, corresponding parts of the aligned sequences showed similar structures. These results are consistent with sites of partial proteolysis of ß[gamma]-crystallin terminal extensions presented here and published previously.
Methods
Experimental part
Expression and purification of recombinant crystallins. Recombinant mouse ßA3- and ßB2-crystallins were expressed using the baculovirus system. Briefly, transfer plasmid pBBßA3 [7] and pBBßB2 [14] were cotransfected with linearized AcMNPV DNA into S. frugiperda Sf9 cells and the recombinant (gal+, occ-) virus plaques were purified as previously described [7].
For expression of the recombinant ßA3- and ßB2-crystallins (rßA3 and rßB2), Sf9 cells were infected with the corresponding purified recombinant virus, harvested, and lysed as described [7]. Both rßA3 and rßB2 were purified from soluble extracts of infected Sf9 cells by anion exchange on a DE52 column followed by gel filtration chromatography on a Superose 75 column [7]. Purity was checked by SDS-PAGE and judged to be 95% or greater by densitometry. Purified rßA3 and rßB2 crystallins were isolated in the absence of protease inhibitors and partially degraded crystallins were resolved by SDS-PAGE and electroblotted to PVDF membrane. Bands were excised and sequenced by the Edman degradation method on Applied Biosystems 470A and 474A sequencers with online PTH-AA analyzer. Similar results were obtained by storing Sf9 lysates at -20 °C with multiple freeze-thaw cycles and by incubating the supernatant at room temperature in the absence of protease inhibitors.
Characterization of the Sf9 protease. Soluble extracts of rßB2-crystallin were used for testing the effect of different protease inhibitors including aprotinin, pepstatin, leupeptin, 4-(2-Amynoethyl)-benzenesulfonyl-fluoride hydrochloride (AEBSF), bestatin and E-64 (Boehringer-Mannheim). These inhibitors were applied with final concentrations of 0.3 µM, 1µM, 1µM, 200 µM, 180 µM and 2.8 µM, respectively. Each inhibitor was applied separately to 200 µl aliquots of the rßB2-crystallin extracts at room temperature in 50 mM Tris-HCl, pH 8.5, 1 mM EDTA, 1 mM DTT, and at 0, 1, 4 and 24 hours aliquots were analyzed on a 12% acrylamide SDS-PAGE gel.
Calpain cleavage sites. The m-calpain induced cleavage sites in the amino-terminal extensions of ß-crystallins were determined by incubating total soluble proteins from the lens of each species with purified m-calpain, separating the resulting digests by two-dimensional electrophoresis, and sequencing partially degraded ß-crystallins following blotting to polyvinylidene difluoride (PVDF) membranes. The m-calpain cleavage sites in the amino-terminus of rat ßA3- and ßB2-crystallins [11], and bovine ßA3- and ßB2-crystallins [15] were previously reported. The previously unpublished m-calpain cleavage sites in human ßA3- and ßB2-crystallins and chicken ßB2-crystallin were similarly determined (David et al., unpublished data).
ß-crystallin sequences are numbered so that the first amino acid residue of the N-terminal core domain is labeled residue 1, with residues increasing in number through both domains and the C-terminal arm. Amino acid residues of the N-terminal arm are numbered in decreasing order from residue -1, adjacent to the core domain, to the N-terminal residue.
Theoretical background
Sequence alignment and protein structure. Sequences of 5 ßA3-crystallins from human (GenBank accession numbers M14301, M14302, M14303, M14304, M14305, M14306), rat (GenBank accession number AF013248), mouse, bovine and chicken [16], and 5 ßB2-crystallin sequences from mouse [17] corrected as described [14], human [17], bovine, rat and chicken [18-20] were aligned using the multiple alignment procedure of Rost & Sander [12]. ßB2-Crystallin structure was taken from the file 1blb from the January 1995 Release of the Brookhaven Protein Data Bank [21]. File 1blb contained four molecules A, B, C and D grouped as two dimers, AB and CD.
Profile of tripeptide usage. We developed a simplified version of the nearest-neighbor algorithm for estimating the occurrence of the residue secondary structure states. The SCAN3D database, incorporated in the WhatIf program, allowed searching of secondary structure content for sequence segments in the database of 308 proteins with known 3D structure [22]. Construction of the profile of tripeptide usage was based on decomposition of the sequence terminal extensions into overlapped tripeptides. For each tripeptide in the terminal arms, 3-D structures in the SCAN3D database were searched, yielding a list of matching tripeptide sequences with secondary structure content for each residue in the tripeptide.
The program assigned one of five conformational states to each amino acid residue, termed Rj, with R being the name of the residue in the j position of the sequence. Each residue Rj has a conformational state Sj. Five conformational states were considered: Sj = Pk, k=1,2,3,4,5, where P1 is a 310-helix, P2 is an [alpha]-helix, P3 is a ß-conformation, P4 is a turn, and P5 includes all other conformations. For example, in any five - residue fragment Rj-2 - Rj-1 - Rj - Rj+1 - Rj+2 of protein sequence three overlapped tripeptides were selected: Rj-2 - Rj-1 - Rj; Rj-1 - Rj - Rj+1; and Rj - Rj+1 - Rj+2. The residue Rj is included in each of these tripeptides in the third, second and first positions, respectively. Thus the conformational state for each residue Rj was ascertained a total of three times, once as part of each of three overlapped tripeptides. Each sequence tripeptide was searched in the SCAN3D database containing proteins of known conformation. For each tripeptide, the conformation states from tripeptides with sequences identical to that under consideration were counted. The number of times each residue Rj from the tripeptide was associated with a specific conformational state Sj in the database was calculated by the formula
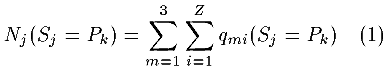
Here m identifies the overlapped tripeptides (m = 1, 2, 3 are the three overlapping peptides in which Rj is the third, second and first residue respectively), Z is the number of identical tripeptides found in the SCAN3D database and qmi (Sj = Pk) is the match for the residue conformation Sj = Pk under consideration for the overlapping tripeptide and the known conformation of the peptide in the database: qmi = 1, if the conformations are identical, and qmi = 0 otherwise.
The observed frequency of the Pk - state occurrence for j-th residue in the sequence is
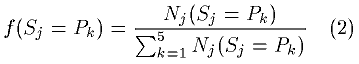
The informational entropy for the residue, which is similar to the estimate of its apparent conformational entropy, was calculated as follows:
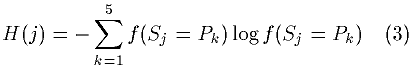
The informational entropy will be higher for residues with relatively equal frequencies for each of the five conformational states and will be lower for residues with marked occurrence preferences for one or a few states.
Finally, the observed frequencies for all five conformational states were grouped into frequencies of three general types of conformations: H - helical (310- and [alpha]-helical), E - extended (ß-sheet) and L - random coil (turns plus all other conformations), respectively. By this method a profile of occurrence frequencies for the tripeptide usage was constructed and compared with the profiles of the secondary structure obtained by the nearest-neighbor algorithm [13] and the PHD prediction program based on multiple alignments [12].
Results
Prediction of the secondary structure of mouse ßA3 - and ßB2-crystallin terminal extensions by overlapping tripeptides
Frequency estimations for two of the three states (H and L for helix and coil, respectively) calculated for each residue as described in the Methods section are presented in Table 1 and Table 2 for ßA3- and ßB2-crystallins, respectively. Helical structures are predicted (>50% frequency) for residues from -20 thorough -24 and from -12 thorough -13 in ßA3-crystallin (Table 1). Residues from -9 to -11 in the N-terminal arm of ßB2-crystallin were also predicted to have a helical conformation (Table 2), although this prediction is less secure since only two matches were seen with tripeptides containing residues -10 and -11. Turns also were predicted for residue T-10 in ßA3- and residues D-13, G-7, W175 and H182 in ßB2-crystallin with a 50% threshold.
The strength of the structural predictions varied for different residues, with tripeptides showing a large number of hits producing statistically more significant results. The most significant tripeptide occurrences were 32 hits for the REL- and 38 hits for the GSL-tripeptides in ßA3-crystallin, and 34 hits for the AGK-tripeptide in ßB2-crystallin (Table 1 and Table 2). For the REL-tripeptide, nearest-neighbor analysis gave a helical conformation with information entropy of about 1.52±0.05, providing one of the strongest predictions seen in the terminal extensions. The GSL-tripeptide was not predicted to assume a preferred conformation, since the highest fraction (coil) is only 0.44 and the entropy estimate (2.1 average) is above the average for the arm (1.89, Table 1). The AGK-tripeptide, with informational entropy of 1.45 is strongly predicted to reside in the L-conformation and have a tendency to form a turn. In general, the residue tripeptides from the C-terminal extension of ßB2-crystallin were found less frequently in the SCAN3D database than those in the N-terminal extension (Table 2).
Secondary structure prediction for ßA3- and ßB2-crystallin terminal extensions by the PHD and nearest neighbor algorithms
Secondary structure prediction was also carried out by the PHD method using multiple sequence alignments, profile analysis, and neural networks [12]. N-terminal sequences of 28 and 30 residues from ßA3- and ßB2-crystallins of five species were used for secondary structure prediction. As shown in Table 3 and Table 4, the method predicted helical and coil conformations for one helical and 7 coil segments with reliability values equal to or greater than 7, indicating an estimated 91.1% reliability.
For ßA3-crystallin the PHD method predicts a helical segment extending from residue E-20 to V-25, and three coil segments extending from E-29 to Q-27, from L-18 to P-17, and from T-10 to L-3. The PHD method did not unambiguously predict a helical or extended conformation in the segment from T-15 thorough T-10, in which the triplet usage predicted two residues in helical conformation. Two coil segments including residues from M-16 to S-14 and from T-10 to N-1 were predicted for the N-terminal extension of ßB2-crystallin (Table 4). Two coil segments from residues G179 to F181 and residues P183 to S185 were also predicted for the C-terminus of ßB2-crystallin.
Residues predicted to participate in helical structures by the nearest-neighbor algorithm NNSSP [13] are also shown in Table 3, Table 4, and Figure 1. Predictions are similar for 4 of the 5 species analyzed. This method predicts a helical conformation for the amino-termini in human, bovine, mouse and rat ßA3-crystallin sequences. For the chicken sequence this method only predicted a helical conformation for two residues. The NNSSP method did not predict any significant helical content in the ßB2-crystallin terminal extensions.
Inverse alignment of ßB2-crystallin terminal extensions
Secondary structure predictions for the amino- and carboxy-terminal arms of ßB2-crystallin were compared with the sequences aligned inversely, similar to their alignment in the crystallographic dimer structure (Figure 2). Thus, the sequence of the N-terminal arm extending from the N-terminal residue to the beginning of the first domain is aligned with the sequence of the C-terminal arm, extending from the C-terminus to the end of the second domain of the molecule.
It can be seen that when they are aligned in opposite orientations, the two terminal extensions of mouse ßB2-crystallin have a homologous segment (Figure 2a) with similar physical properties, such as hydrophobicity and hydrophilicity, for corresponding residues. This alignment extends from residues A-8 to N-1 in the amino-terminal arm, corresponding to the residues from A180 to M173 in the C-terminal arm. The remaining residues of both arms, residing at the N- and C-termini, have unlike sequences. Residues from S-14 to Q-9 in the N-terminus are hydrophilic while the C-terminal residues from P183 to F181 are hydrophobic.
The central part of each terminal arm contains the tripeptide AG(K/R) made up of the hydrophobic residue Ala, a central glycine and positively charged Lys or Arg in the third position of the alignment (Figure 2a). The AG(K/R) tripeptide occurs relatively frequently (34 hits) in the SCAN3D database compared to other tripeptides (Table 2). The AGK-tripeptide has relatively low informational entropy in the amino-terminal extension (1.45, see Table 2). Although this tripeptide is most often seen in a loop conformation, glycine is known to be able to assume a wide range of torsion angles, suggesting that this tripeptide might be highly flexible.
Similar alignments for the N- and C-terminal arms of ßB2-crystallin from five different species are presented in Figure 2b,c. All five sequences have a high homology so that the alignment derived for the mouse sequence also holds for the other four ßB2-crystallins with the exception that the central Gly-residue is replaced by Ser in the chicken amino-terminal arm. Thus, both terminal extensions of ßB2-crystallin appear to be separated into two distinct segments connected by a flexible AG(K/R) tripeptide. Amino-terminal residues Q-11, T-10 and Q-9 beyond the flexible tripeptide are predicted to have a helical conformation.
Results of limited proteolysis
When recombinant mouse ßA3- and ßB2-crystallins are expressed in Sf9 cells they both are susceptible to truncation of the amino-terminal arms unless protease inhibitors are included during purification. The recombinant mouse ßA3-crystallin truncation site was characterized by Hope et al. [7] and is shown in Figure 1. This cleavage site is located between residues -23 and -24, close to the beginning of the helical segment. Native ßB2-crystallin begins to show truncation after 1 hour of incubation at room temperature in absence of a protease inhibitor (Figure 3b) and is completely truncated after 4 hours. Intact and truncated ßB2-crystallins are represented on the SDS-PAGE gel by two different bands of MW 25.7 and 24.3 kDa, respectively. The protease activity in the Sf9 cell extracts was characterized with respect to its sensitivity to inhibitors: it is inhibited by leupeptin and E-64, but is insensitive to AEBSF and bestatin. These results are consistent with the presence of a thiol protease in Sf9 extracts. N-Terminal sequence analysis of the 24.3 kDa band indicates the presence of two fragments with primary sequences identical to ßB2-crystallin: the first (55% of signal) begins immediately before residue A-8, while the second (45% of total signal) begins immediately after A-8, one residue internal to the first. The cleavage site before residue A-8 corresponds to the position expected to be cut by a thiol protease from the inverse alignment, where it is seen to be the first residue in the AGK triplet (Figure 2), and from structural predictions based on tripeptide usage (Figure 1).
Other proteolytic cleavage sites identified in ß-crystallin arms have been published previously for different species as detailed in the caption to Figure 1. The cleavage sites are superimposed on the terminal sequences with the conformations predicted by the tripeptide usage, the nearest-neighbor [13] and PHD algorithms [12]. In the rat [11,23,24], human [25; David, et al., unpublished data] and bovine [15] amino-terminal extensions of ßA3-crystallin, the major m-calpain cleavage sites are located between residues -19 and -20 just at the end of predicted helical segments. Minor m-calpain cleavage sites are also located near residues with predicted helical or turn conformations. The cleavage site occurring naturally in the rat lens ßA3-crystallin terminal extensions coincides with the major m-calpain cleavage site at T-19, adjacent to the predicted helical segment. In the bovine ßA3-terminal extension [15], the natural cleavage site is located one residue from T-9, which has a predicted turn conformation. In the chicken ßA3-terminal extension no m-calpain cleavage is detected (David et al., unpublished data), and these results correlate with predictions of the tripeptide usage method which do not show a helical conformation for segment -20 to -25, in contrast with results of the rat, human and bovine sequences. However, a naturally occurring cleavage site in chicken ßA3-crystallin is located one residue from the predicted M-13 to T-10 helical segment.
Cleavage sites in the rat and mouse ßB2-crystallin terminal extensions [24] are located following the predicted helical fragment, between that segment and the last residue of the homologous fragment from N-1 to A-8 (Figure 1 and Figure 2a). Similar results were also obtained for bovine and human ßB2-crystallin terminal extensions (sequences not shown). In the chicken sequence the major m-calpain cleavage site (David et al., unpublished data) is located 3 residues from a predicted helical fragment, just before the ASK tripeptide. The naturally occurring cleavage site is located one residue from the predicted helical fragment.
Discussion
Structure predictions using two versions of modified nearest-neighbor algorithms and the PHD prediction method all predict the possible location of structural microdomains in the amino- and carboxy-terminal arms of the ß-crystallin. The predicted microdomains are consistent among the three methods used, and are also consistent in most cases with the results of limited proteolysis of representative ß-crystallins using two proteases. Nearest-neighbor algorithms predict secondary structure for globular proteins with accuracy 64-68% for single sequences and above 70% when using evolutionary conformation.
Here, a modification of the nearest-neighbor algorithm based on comparison of overlapping tripeptides rather than longer sequences was used. This modification was based on crystallographic and NMR data showing that the terminal extensions are mobile and exposed in solvent [5,26], consistent with the absence of stable long-range interactions in the terminal extensions. This suggests that their secondary structure might be predicted best by considering only short-range interactions between residues in the arm. Although 5 residues are required to form one turn of helix, we also included segments containing one or a few residues predicted to have a high propensity to assume a helical conformation. This is because the preferred state of a residue, i, is a property not only of that residue but of the 5 residues surrounding it, i-2 thorough i+2 (see Methods). The coil (L) conformation predicted by this method for amino acids A-8 thorough N-1, Q174 and W175 of ßB2-crystallin and shown in Table 2 is consistent with the crystal structure [4].
The PHD method predicts structure using a different approach based on mutation profile analysis and neural network algorithms [12]. The tripeptide usage and nearest-neighbor algorithms [13] predict a helical amino-terminal segment in the amino-terminal extension of ßA3- and ßB2-crystallins in most species, with the exception of the chicken (Figure 1). However in this case the tripeptide usage predicted a helical conformation for four residues located very close to the endogenous truncation site.
The alignment of the amino- and carboxy-terminal extensions shown in Figure 2 is suggested by the crystallographic structure of bovine ßB2-crystallin [4,26], in which the C-terminal extension of one member of a dimer pair and the N-terminal extension of the other are closely positioned (Figure 4). As shown in this figure, the C[alpha]-C[alpha] distance between residue W175 situated at the C-terminus of the first molecule in the dimer and Leu-2 at the N-terminus of the second molecule is 0.517 nm for the first and 0.527 nm for the second dimer in the 1blb file. These residues are sufficiently close to suggest that residues W175 and L-2 are interacting, suggesting that the terminal extensions can be aligned starting from these residues with the arms superimposed in opposite orientations as shown in Figure 2.
The secondary structure predictions summarized in Figure 1 are supported by the patterns of proteolytic degradation seen in the terminal arms. The histogram in Figure 5 shows the number of cleavages occurring at given distances from the end of the nearest helical segment. Positions for 8 out of total 18 cleavages correlate precisely with the ending or beginning of helical segments with 5 additional cleavages located 1 residue off, demonstrating a strong tendency of these proteases to cut a sequence adjacent to compact helical segments. This hypothesis was tested statistically using data from Figure 1. Peptide bonds in the terminal arms were divided into two groups. The first group consists of those between the terminal residue of a predicted helical segment and the adjacent residue not belonging to that helical segment. The second group consists of all remaining peptide bonds in the terminal arm including bonds between non-terminal residues within predicted helical segments and those between residues lying outside of helical segments. Cleavage of bonds adjacent to predicted helical segments was highly favored ([chi]2 = 119.9, 1 df, p < 0.001).
Our results agree with data showing that a conformational factor is involved in mechanism of action for µ-calpain and m-calpain, and that both enzymes may recognize certain conformations of substrate molecules [27]. In the current model of vimentin structure [28] µ-calpain acts primarily on non [alpha]-helical regions and this model agrees with recently published results that the µ-calpain cleavage site of [alpha]II spectrin occurs in the exposed loop juxtaposed between helix C of the spectrin repeat and the calmodulin binding domain [29].
However, cleavage sites were observed in only 62% of all positions adjacent to predicted helical segments. Relative sequence specificity of the thiol proteases in the lens and baculovirus, or the relatively small number of cleavages studied may explain the lack of observed cleavages at the other 38% of predicted sites. In addition, the algorithms used in this study to predict helical segments are imperfect. This includes the prediction of precise segment ends using overlapping tripeptides whose predicted stated is dependent on five adjacent amino acids. Cleavage sites detailed in Figure 1 show no obvious sequence similarity, consistent with previous observations that the thiol proteases are relatively non-specific in their cleavage sites. While the site specificity of µ- and m-calpains deduced from peptide cleavage studies is not very rigid, these proteases seem to avoid cleavage at proline residues and to favor cleavage adjacent to sites of the form X-Z, where X is leucine, valine or isoleucine and Z is tyrosine, arginine, or lysine [27]. No such sites are seen in any of the terminal arm sequences in this study, and of the 16 cleavage sites only 6 (including 4 structurally predicted sites) have a single amino acid match with the preferred sequence. It is interesting to note that in the bovine ßA3-crystallin amino-terminal arm, which contains the only calpain cleavage site 2 residues within a predicted helical segment, the helical region ends in a proline, which might both disrupt the helical structure and discourage calpain cleavage precisely at the terminus. However, in cleavage of most proteins by calpain, the amino acid pattern predicted from peptide studies does not tend to be followed well. In studied proteins, an open or nonhelical conformation or location at the boundary between hydrophilic and hydrophobic clusters seems to favor cleavage [27].
The occurence of similar calpain cleavage sites in the terminal sequences of ß-crystallins from different species and in insect cell culture suggests that the structural properties of the terminal extensions might provide a common element for recognition by these proteases. The pattern of proteolysis for both, ßA3- and ßB2-crystallin terminal extensions can be explained by the presence of segments with a local secondary structure, which might be thought of as microdomain structures. While these structures are not stable or constant enough to appear on crystallographic maps, they represent a favored conformation for the terminal extension. Proteolytic cleavage sites in the N-terminal arms are located mainly at the beginning or the end of predicted helical structures, and also may occur near residues in a turn conformation. Experiments to explore this hypothesis further by NMR and CD analysis of synthetic oligopeptides are underway, although it is unclear whether putative microdomain structures sufficient to affect protease activity would be detectable in this fashion.
The importance of proteolysis in formation of the selenite induced cataract was demonstrated by Shearer et al. [9]. Cleavage of the N-terminal extensions of ßA3- and/or ßB2-crystallin has been demonstrated by Takemoto et al. [30], David et al. [11,23,24], Hope et al. [7] and in the present study. These results show that the amino-terminal arms of ß-crystallins are highly accessible to thiol proteases like m-calpain or that found in Sf9 cells. These data correlate well with crystallographic and NMR data indicating that the N-terminal extension of ßB2-crystallin is freely located in the solvent without stable interactions with structural domains [5,26]. However, at least part of the carboxy-terminal arm appears to be important for structural domain binding and swapping [6].
In summary, we suggest a simple structural model for the ß-crystallin terminal extensions. For each terminal arm considered here, segments with a homogeneous secondary structure can be predicted. The helical microdomain follows the first coil segment in the N-termini of the ßA3- and ßB2-crystallins. These segments contain 4 or 3 polar residues with negative charges and contribute to the net negative charge of the N-termini. Next, two coiled segments of the N-terminal extension of ßA3-crystallin follow the helical segment, and are separated by a short segment with a tendency for helical (or extended as predicted by PHD) conformation. The last coil segment in both crystallins contains four or five hydrophobic residues and will have some tendency to participate in hydrophobic interactions. Possibly, this segment may interact with a common part of the C-terminal segment in ßB2-crystallin (Figure 2).
If the terminal extensions of the ß-crystallins do indeed form microdomain structures, size estimations for terminal extensions might be reconsidered. Recently Zarina et al. [31] predicted the structure of [gamma]S-crystallin. The structure of the N-terminal extension was modeled as an extended chain so that it did not interact with the body of protein. However, if the terminal arms of the ß-crystallins assume an extended random conformation it would seem likely that they must contact and interact with crystallins or other constituents of the lens cell given the high protein concentrations. Indeed, terminal extensions of the bovine acidic ß-crystallins are 13, 11, 30 and 11 residues long for the ßA1-, ßA2-, ßA3-, and ßA4-crystallins, respectively. Basic ß-crystallins have N-terminal extensions of 59, 16, and 23 residues, and C-terminal extensions of 17, 13, and 13 residues in ßB1-, ßB2-, and ßB3-crystallins, respectively [32]. If they assume an extended conformation the length of N-terminal extensions would range from 3.5 nm to 9.6 nm for acidic, and from 5.1 nm to 18.8 nm for basic crystallins. Similarly, The C-terminal extension of basic crystallins would range from 4.1 nm to 5.1 nm in length. By comparison, the radius of each globular domain in the ß[gamma]-crystallins is about 1.5 nm and the largest dimension in the ßB2-crystallin molecule is about 6.6 nm as measured in the 3D model of this protein (1blb file of PDB). Thus, if terminal extensions assume an extended conformation, the dimension of the terminal arm would be comparable to or greater than the domain size. One alternative is that elements of secondary structure or small microdomain regions with relatively compact structure may exist in the N-terminal arms of ß-crystallins, reducing the estimate for the real size of terminal extensions.
Acknowledgements
This work was funded in part by R01 EY12016 from the National Eye Institute.
References
1. Slingsby C, Driessen HP, Mahadevan D, Bax B, Blundell TL.
Evolutionary and functional relationships between the basic and acidic
beta-crystallins. Exp Eye Res 1988; 46:375-403. ![]()
2. den Dunnen JT, Moormann RJ, Schoenmakers JG. Rat lens
beta-crystallins are internally duplicated and homologous to
gamma-crystallins. Biochim Biophys Acta 1985; 824:295-303.
![]()
3. Berbers GA, Hoekman WA, Bloemendal H, de Jong WW, Kleinschmidt T,
Braunitzer G. Homology between the primary structures of the major
bovine beta-crystallin chains. Eur J Biochem 1984; 139:467-479.
![]()
4. Nalini V, Bax B, Driessen H, Moss DS, Lindley PF, Slingsby C.
Close packing of an oligomeric eye lens beta-crystallin induces loss of
symmetry and ordering of sequence extensions. J Mol Biol 1994;
236:1250-1258. ![]()
5. Carver JA, Cooper PG, Truscott RJ. 1H-NMR spectroscopy of beta
B2-crystallin from bovine eye lens. Conformation of the N- and
C-terminal extensions. Eur J Biochem 1993; 213:313-320.
![]()
6. Sergeev YV, Hejtmancik JF. A method for determining domain binding sites in proteins with swapped domains: implications for betaA3- and betaB2-crystallins. In: Marshak DR, editor. Techniques in Protein Chemistry, Vol. VIII. New York: Academic Press; 1997. p. 817-826.
7. Hope JN, Chen HC, Hejtmancik JF. BetaA3/A1-crystallin association:
role of the N terminal arm. Protein Eng 1994; 7:445-451.
![]()
8. Trinkl S, Glockshuber R, Jaenicke R. Dimerization of beta
B2-crystallin: the role of the linker peptide and the N- and C-terminal
extensions. Protein Sci 1994; 3:1392-1400. ![]()
9. Shearer TR, Ma H, Fukiage C, Azuma M. Selenite nuclear cataract:
review of the model. Mol Vis 1997; 3:8 <http://www.molvis.org/molvis/v3/p8/>. ![]()
10. Norledge BV, Mayr EM, Glockshuber R, Bateman OA, Slingsby C,
Jaenicke R, Driessen HP. The X-ray structures of two mutant crystallin
domains shed light on the evolution of multi-domain proteins. Nat Struct
Biol 1996; 3:267-274. ![]()
11. David LL, Shearer TR. Beta-crystallins insolubilized by calpain
II in vitro contain cleavage sites similar to beta-crystallins
insolubilized during cataract. FEBS Lett 1993; 324:265-270.
![]()
12. Rost B, Sander C. Combining evolutionary information and neural
networks to predict protein secondary structure. Proteins 1994;
19:55-72. ![]()
13. Salamov AA, Solovyev VV. Prediction of protein secondary
structure by combining nearest-neighbor algorithms and multiple sequence
alignments. J Mol Biol 1995; 247:11-15. ![]()
14. Hejtmancik JF, Wingfield PT, Chambers C, Rusell P, Chen HC,
Sergeev YV, Hope JN. Association properties of betaB2- and
betaA3-crystallin: ability to form dimers. Protein Eng 1997;
10:1347-1352. ![]()
15. Shih M, Lampi KJ, Shearer TR, David LL. Cleavage of
beta-crystallins dring maturation of bovine lens. Mol Vis 1998; 4:4 <http://www.molvis.org/molvis/v4/p4/>. ![]()
16. van Rens GL, de Jong WW, Bloemendal H. A superfamily in the
mammalian eye lens: the beta/gamma-crystallins. Mol Biol Rep 1992;
16:1-10. ![]()
17. Chambers C, Russell P. Sequence of the human lens
betaB2-crystallin-encoding cDNA. Gene 1993; 133:295-299.
![]()
18. Hogg D, Gorin MB, Heinzmann C, Zollman S, Mohandas T, Klisak I,
Sparkes RS, Breitman M, Tsui LC, Horwitz J. Nucleotide sequence for the
cDNA of the bovine beta B2 crystallin and assignment of the orthologous
human locus to chromosome 22. Curr Eye Res 1987; 6:1335-1342.
![]()
19. Aarts HJ, Lubsen NH, Schoenmakers JG. Crystallin gene expression
during rat lens development. Eur J Biochem 1989; 183:31-36.
![]()
20. Duncan MK, Banerjee-Basu S, McDermott JB, Piatugorsky J. Sequence
and expression of chicken betaA2- and betaB3 crystallins. Exp Eye Res
1996; 62:721-722. ![]()
21. Abola E, Bernstein FC, Bryant SH, Koetzle TF, Weng J. Protein data bank. In: Allen FH, Bergerhoff G, Sievers R, editors. Crystallographic databases-information content, software systems, scientific applications. Data Commission of the International Union of Crystallography. Cambridge 1987; p. 107-132.
22. Vriend G, Sander C, Stouten PF. A novel search method for protein
sequence--structure relations using property profiles. Protein Eng 1994;
7:23-29. ![]()
23. David LL, Shearer TR, Shih M. Sequence analysis of lens
beta-crystallins suggests involvement of calpain in cataract formation.
J Biol Chem 1993; 268:1937-1940. ![]()
24. David LL, Azuma M, Shearer TR. Cataract and the acceleration of
calpain-induced beta-crystallin insolubilization occurring during normal
maturation of rat lens. Invest Ophthalmol Vis Sci 1994; 35:785-793.
![]()
25. Lampi KJ, Ma Z, Shih M, Shearer TR, Smith JB, Smith DL, David LL.
Sequence analysis of betaA3, betaB3, and betaA4 crystallins completes
the identification of the major proteins in young human lens. J Biol
Chem 1997; 272:2268-2275. ![]()
26. Lapatto R, Nalini V, Bax B, Driessen H, Lindley PF, Blundell TL,
Slingsby C. High resolution structure of an oligomeric eye lens
beta-crystallin: Loops, arches, linkers and interfaces in betaB2 dimer
compared to a monomeric gamma-crystallin. J Mol Biol 1991;
222:1067-1083. ![]()
27. Takahashi K. Calpain Substrate Specificity. In: Mellgren RL, Murachi T, editors. Intracellular Calcium-dependent Proteolysis. Boca Raton (FL): CRC Press; 1990. p. 55-74.
28. Geisler N, Weber K. The amino acid sequence of chicken muscle
desmin provides a common structural model for intermediate filament
proteins. EMBO J 1982; 1:649-1656. ![]()
29. Stabach PR, Cianci CD, Glantz SB, Zhang Z, Morrow JS.
Site-directed mutagenesis of alpha II spectrin at codon 1175 modulates
its mu-calpain susceptibility. Biochemistry 1997; 36:57-65.
![]()
30. Takemoto L, Takemoto D, Brown G, Takehana M, Smith J, Horwitz J.
Cleavage from the N-terminal region of beta Bp crystallin during aging
of the human lens. Exp Eye Res 1987; 45:385-392. ![]()
31. Zarina S, Slingsby C, Jaenicke R, Zaidi ZH, Driessen H,
Srinivasan N. Three-dimensional model and quaternary structure of the
human eye lens protein gamma S-crystallin based on beta- and
gamma-crystallin X-ray coordinates and ultracentrifugation. Protein Sci
1994; 3:1840-1846. ![]()
32. Slingsby C, Bateman OA. Quaternary interactions in eye lens
beta-crystallins: basic and acidic subunits of beta-crystallins favor
heterologous association. Biochemistry 1990; 29:6592-6599.
![]()