 Molecular
Vision 2012; 18:2153-2164
<http://www.molvis.org/molvis/v18/a227>
Molecular
Vision 2012; 18:2153-2164
<http://www.molvis.org/molvis/v18/a227> Received 19 December 2011 | Accepted 27 July 2012 | Published 1 August 2012
Molecular
Vision 2012; 18:2153-2164
<http://www.molvis.org/molvis/v18/a227>
Received 19 December 2011 | Accepted 27 July 2012 | Published 1
August 2012
M. Amadio,1 C. Osera,1 G. Lupo,2 C. Motta,2 F. Drago,3 S. Govoni,1 A. Pascale1
1Department of Drug Sciences, Section of Pharmacology, University of Pavia, Pavia, Italy; 2Department of Biochemistry, University of Catania, Catania, Italy; 3Department of Clinical and Experimental Biomedicine, Section of Pharmacology and Biochemistry, University of Catania, Catania, Italy
Correspondence to: Marialaura Amadio, Department of Drug Sciences, Section of Pharmacology, University of Pavia, Viale Taramelli 14, 27100 Pavia, Italy; Phone: +39-0382-987888; FAX: +39-0382-987405; email: amadio@unipv.it
Purpose: To explore whether, following direct contact, there is mutual influence between pericytes (PC) and endothelial cells (EC), and to establish whether protein kinase C (PKC) activation, a condition associated with hyperglycemia, can affect, via the mRNA-binding Hu-antigen R (HuR)/ELAV protein, the expression of vascular endothelial growth factor (VEGF).
Methods: PC and EC were cultured separately or in direct contact (1:1 ratio), and exposed or not to phorbol esters, a PKC activator (100 nM for 15 min). Barrier integrity was evaluated by measuring endothelial electrical resistance and permeability to sodium fluorescein. Immunocytochemistry was performed to visualize EC and PC in coculture, and to evaluate phorbol 12-myristate-13-acetate (PMA)-induced HuR translocation. PKCβI/βII, HuR, and VEGF protein content was measured with western blotting, VEGF secretion in cell culture medium was evaluated with enzyme-linked immunosorbent assay (ELISA), and quantification of VEGF mRNA was performed with real-time quantitative PCR.
Results: In monocultures, VEGF mRNA/protein basal levels were more elevated in PC than in EC. However, the basal expression of VEGF protein, but not mRNA, in PC and EC was affected by culture conditions. In fact, physical contact with PC upregulated VEGF protein levels in the EC, while VEGF was downregulated in PC cocultured with EC. In this last condition, PKCβII and HuR protein basal levels were also decreased in monocultured PC. Moreover, in basal conditions, the amount of VEGF released from the coculture was higher than from the monocultures. Direct activation of PKCβ induced HuR translocation from the nuclear area to the cytoplasm, and increased the protein levels of the kinase itself, HuR, and VEGF in PC and EC in both culture conditions. Concerning VEGF mRNA, PKC activation induced an increase in PKC levels only in monocultured EC and, conversely, a significant decrease in the same transcript amount in cocultured PC. PMA stimulus also led to a significant increase in VEGF secretion in coculture.
Conclusions: When cocultured with PC, EC form a significantly tighter barrier than the endothelial monolayer. The physical contact leads to opposite changes in VEGF protein levels in PC and EC. In particular, in basal conditions, cocultured PC seemed to downregulate their own expression of this proproliferating factor, as well as that of PKCβII and HuR, likely to maintain the 1:1 ratio with the cohabiting EC. In mono- and cocultured PC/EC, PKC direct activation led to a similar increase in PKCβI/βII, HuR, and VEGF protein levels, changes that may also occur at early stages of diabetic retinopathy. The release of VEGF in the medium was favored by physical contact between PC and EC and was further increased by PMA exposure. In contrast with the effects on VEGF protein, PKCβ activation induced modifications in VEGF mRNA content that are different in function of the cell type and the culture conditions. These findings suggest that the changes in the VEGF protein and transcript observed in PC/EC can be ascribed to distinct and concomitant pathways. Further studies on this in vitro coculture model would be useful to better understand the PC/EC interaction in physiologic and pathological conditions.
Diabetic retinopathy accounts for about 5% of cases of blindness worldwide, making diabetes the leading cause of vision loss in working-age adults. Although in some cases diabetic retinopathy signs are localized in the central macula and symptoms may be observed at early stages, the majority of patients with diabetic retinopathy are asymptomatic until the late stages of the disease [1], emphasizing the need to identify early markers of the pathology for an effective therapeutic intervention. Within this context, some studies indicate that disturbances in the microvascular function may arise before hyperglycemia and vascular pathological alterations are evident [2]. Among others, an early, consistent, and specific retinal alteration in diabetic retinopathy is the death of pericytes (PC), microvascular contractile cells providing vessel stability and regulating endothelial proliferation [3,4]. PC loss, thickening of the basement membrane, and alterations in blood flow are followed by local hypoxia, vascular endothelial growth factor (VEGF) production, and increased vascular permeability [5]; despite the increased production of VEGF, a potent survival and proliferating factor for endothelial cells (EC), the latter cells degenerate, leading to microcircle dysfunctions. As the disease progresses, vascular alterations, such as microaneurysms and hemorrhages, become evident upon ophthalmoscopic examination, and this stage is classified as non-proliferative diabetic retinopathy. In some patients, the disease progresses to proliferative diabetic retinopathy, characterized by the formation of new blood vessels that are fragile and easily broken, leaking blood into the retina and vitreous and thus compromising vision [6].
Within physio- and pathological vascular contexts, in recent years PC have gained increasing attention as fundamental constituents of blood microvessels. PC are unique due to their wide distribution and close contact with EC, particularly in the retina, where the ratio between these two types of cells is 1:1, the highest in the body [7]. Studies performed in mouse models characterized by inactivation of genes involved in the vascular development and function indicate that an intimate interaction between EC and PC exists to the extent that impairment of one of these two cell types unavoidably affects the other one [3]. However, in spite of the ongoing progress in understanding the endothelial cells/pericytes interaction, this biologic system needs to be better characterized in physiologic and pathological conditions to determine which changes occurring in one of the two cell types do reverberate on the other one.
In retinal vascular cells, such as PC, the early biochemical changes associated with diabetic hyperglycemia lead to the generation of diacylglycerol (DAG [8]), a protein kinase C (PKC) activator. The activation of specific PKC isoforms, mainly βI/II, has been reported as a biochemical player involved in the early stages of diabetic retinopathy development (reviewed in [8]). Indeed, the loss of retinal pericytes and the formation of acellular capillaries have been linked to hyperglycemic activation of PKC [9]. Moreover, in vivo studies provided direct evidence that retinal DAG (or analogs)–induced PKCβ activation can mimic the hemodynamic abnormalities usually observed in diabetic rats [10,11], suggesting that PKC activation is a primary biochemical trigger of the retinal microvascular changes observed in diabetes. PKC activation is, in turn, responsible for the increased expression of several genes encoding for growth factors and proteins involved in vascular permeability, such as VEGF, one of the most important players in maintaining homeostasis in retinal vascular cells [12]. Among the several VEGF isoforms divided into five groups (VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor) that play opposite roles in diabetic retinopathy [13], VEGF-A165 is the most common and important for angiogenesis [14] in physiologic and pathological conditions. The importance of VEGF in ocular diseases is supported by the extensive use of specific anti-VEGF molecules, such as bevacizumab and ranibizumab, in age-related macular degeneration and diabetic retinopathy [15].
Studies conducted by our and other laboratories have shown that in bovine pericytes and human vascular smooth muscle cells VEGF gene expression is regulated by a PKC-dependent mechanism [16,17]. In particular, we demonstrated, in vitro in PC and in vivo in the retina, the existence of a molecular cascade specifically involving PKCβ and the mRNA-binding Hu-antigen R (HuR)/ELAV protein, finally resulting in increased expression of the VEGF protein [16,18].
Numerous studies highlight the importance of the cross-talk among microcapillary cells [19,20]. Within this context, researchers have demonstrated that retinal capillary coverage by pericytes is crucial for the survival of endothelial cells [21], particularly under stress conditions such as diabetes. In the presence of high VEGF levels, such as in the retinopathy of prematurity model, pericyte deficiency leads to reduced inhibition of endothelial proliferation in vivo [22]. Therefore, the in vitro models of the blood–retinal barrier (BRB), which are also involved in studies on diabetic retinopathy, are based on cocultures of retinal microcapillaries, endothelial cells, and pericytes in the presence or not of astrocytes [23]. These cells were shown to maintain specific in vivo BRB properties by expressing high levels of the endothelial junction proteins occludin, claudin-5, VE-cadherin, and Zona occludens protein-1 at cell borders, and the specific pumps glucose transporter-1 and efflux transporter P-glycoprotein, which influence the permeability of the model to differently-sized molecular tracers [24,25]. We employed PC and EC in monocultures and cocultures; the latter represented an in vitro model of BRB. The major aim of the present study was to explore whether, following direct contact of PC and EC, there is mutual influence between these cells; in particular, in monocultured and cocultured PC/EC we wanted to establish whether direct PKC activation, a condition associated with hyperglycemia, can affect, via the mRNA-binding HuR/ELAV protein, the expression of VEGF, thus recapitulating an important biochemical change observed in early stages of diabetic retinopathy.
Primary bovine retinal endothelial cells (EC) were purchased from Sigma (Milan, Italy) and fed with Ham’s F10 medium supplemented with 10% fetal bovine serum (FBS), 80 μg/ml heparin, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Pure microvessel PC cultures were prepared from the bovine retina as previously described [26]. The retinas were homogenized in minimum essential medium, and filtered through an 80 um nylon sieve. The trapped microvessels were digested in phosphate-buffered saline, pH 7.4, containing 1 mg/ml collagenase-dispase and 0.5% bovine serum albumin for 20 min at 37 °C.
The homogenate was subjected to centrifugation (1,000× g, 10 min), and the isolated cells were then cultured in Dulbecco's Modified Essential Medium (DMEM) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Culture plates were previously coated with 2 mg/ml solution of rat tail collagen.
Inserts (Transwells, Corning Inc., Corning, NY) were coated on the upper and bottom sides with 2 mg/ml solution of rat tail collagen (Sigma) containing 10 fold concentrated DMEM plus 0.3 M NaOH. The coating was dried for 1 h at 37 °C and was rinsed twice with water, and once with Ca2+- and Mg2+-free phosphate buffer saline (containing KH2PO4 1.06 mM, NaCl 155.2 mM, Na2HPO4-7H2O 2.97 mM) before being placed in complete medium. To make an in vitro model of the BRB based on the cells’ direct contact, PC were first plated on the outside of the polycarbonate membrane (40,000 cells/cm2) of the Transwell inserts (six-well type, 0.4-μm pore size), and directed upside down in the well culture plate. After the PC had adhered, the Transwells were inverted and reinserted into six-well plates, and EC were plated on the top surface of the insert (40,000 cells/cm2). After coincubation for 24 h, the medium was discarded and replaced with fresh medium (50% DMEM plus 50% F-10 HAM’s containing 10% FBS); incubation with or without 100 nM phorbol 12-myristate-13-acetate (PMA, Sigma) was then performed for 15 min. At the end of the incubation period, cells grown on both sides of the inserts were scraped with a rubber policeman and saved. Since the protein/mRNA yields obtained from the cocultures were low, more than eight treatments of cocultured and monocultured PC/EC cells were used to perform all the experiments.
Trans endothelial electric resistance (TEER) was measured using a Millicell-ERS (Millipore, Milan, Italy). The collagen-treated Transwell inserts were used to measure the background resistance. Values were expressed as ω × cm2 and were calculated with the following formula: [the average resistance of experimental wells - the average resistance of blank wells] × 0.33 (the area of the Transwell membrane). To determine the flux of sodium fluorescein (Na–F) across the endothelial monolayer, inserts containing cell cultures were transferred to 12-well plates containing 1.5 ml Ringer–HEPES buffer (136 mM NaCl, 0.9 mM CaCl2, 0.5 mM MgCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 10 mM NaH2PO4, 25 mM glucose, and 10 mM HEPES, pH 7.4) in the lower or abluminal compartments. In the inserts (luminal compartment), the culture medium was replaced with 0.5 ml buffer containing 10 μg/ml Na–F (MW: 376 Da). The inserts were transferred at 5, 15, and 30 min to a new well containing Ringer–HEPES buffer. The concentrations of the marker molecule in samples from the upper and lower compartments were determined with a fluorescence multiwell plate reader (PerkinElmer, Monza, Italy; excitation wavelength: 485 nm, emission wavelength: 535 nm). Flux across cell-free inserts was also measured, and the trans-endothelial permeability coefficient (Pe) was calculated. Permeability measurements of triplicate filters for each culture condition were performed. Transport was expressed as microliters of donor (luminal) compartment volume from which the tracer is completely cleared:

The average cleared volume was plotted versus time, and permeability x surface area product value for the endothelial monolayer (PSe) was calculated with the following formula:
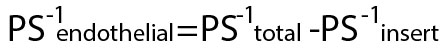
The PSe divided by the surface area (1 cm2 for Transwell-12) generated the endothelial permeability coefficient (Pe in 10−6 cm/s).
To characterize PC and EC in coculture, immunocytochemistry with a confocal-fluorescent microscope was performed. After incubation in coculture, PC or EC grown on one side of the filter were removed by rubbing on filter paper to leave only one cell type. Filters with PC or EC were washed, fixed by adding 4% paraformaldehyde in PBS, and processed for immunocytochemistry as previously described [27]. Briefly, the cells were washed in PBS and preincubated in blocking buffer (0.5% Triton X-100, 5% BSA and 5% donkey serum in PBS) for 20 min at room temperature. The staining was performed by, using the following antibodies (all from Santa Cruz, Santa Cruz, CA): anti-α actin (PC marker) mouse monoclonal antibody, coupled to a fluorescein isothiocyanate–labeled secondary antibody, or anti-von Willebrand factor (vWF; EC marker) rabbit polyclonal antibody, coupled to a red fluorescent-labeled Cy3 secondary antibody, both used to highlight cell architecture; anti-HuR mouse monoclonal antibody, coupled to a red fluorescent-labeled Cy3 secondary antibody, to evaluate HuR cellular localization. Distribution of immunocomplexes was observed with confocal immunofluorescence microscopy using a Leica TCS NT confocal laser scanning microscope (Leica Microsystems, Milan, Italy). Single lower power scans were followed by 16–22 serial optical sections of randomly chosen cells in four to five fields per coverslip.
Cells were homogenized in a buffer containing (20 mM Tris [pH 7.4], 2 mM EDTA, 0.5 mM ethylene glycol tetraacetic acid (EGTA), 50 mM 2-mercaptoethanol, 0.32 mM sucrose, and a protease inhibitor cocktail [Roche Molecular Biochemicals, Mannheim, Germany] at the dilution suggested by the manufacturer) by using a Teflon/glass homogenizer. Proteins were measured according to the Bradford method using bovine albumin as a standard. Proteins were diluted in 2× sodium dodecyl sulfate protein gel loading solution, boiled for 5 min, separated on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The samples were transferred onto a nitrocellulose membrane which was exposed to an horseradish peroxidase-conjugated secondary antibody. The nitrocellulose membranes signals were detected by chemiluminescence previously described [28]. The anti-PKCβI mouse monoclonal antibody (sc-8049; Santa Cruz) was diluted at 1:300, the anti-PKCβII rabbit polyclonal antibody (sc-210; Santa Cruz) at 1:150, the anti-HuR mouse monoclonal antibody (sc-5261; Santa Cruz) at 1:1000, the anti-VEGF rabbit polyclonal antibody (sc-507; Santa Cruz, recognizing VEGF165/121/189) at 1:1,000, and the mouse monoclonal anti-α-tubulin (T9026; Sigma) at 1:1,000. The secondary antibodies were diluted at 1:3,000. The nitrocellulose membrane signals were detected with chemiluminescence. Experiments were performed at least three times for each different cell preparation, and α-tubulin was used to normalize the data. Statistical analysis of western blot data was performed on the densitometric values obtained with the NIH image software 1.61.
Total RNA was extracted from cells using the RNeasy Micro Plus Kit (Qiagen, Milan, Italy). The reverse transcription was performed following standard procedures. PCR amplifications were performed using the LightCycler instrument (Roche Molecular Biochemicals) in the presence of QuantiTect SYBR Green PCR mix (Qiagen) and the specific primers for VEGF (forward: TCA CCA AAG CCA GCA CAT AG; reverse: AAC AAG GCT CAC AGT GAT TTT CT; melting temperature: 59 °C, product size: 107 bp) [16]. The ribosomal protein L10a (RPL10a) mRNA was chosen as the reference mRNA on which VEGF was normalized because this RNA remained substantially stable during the treatments (data not shown).
VEGF-A protein released in the medium from the different cell cultures was estimated with an enzyme-linked immunosorbent assay (ELISA) kit (Bethyl Laboratories, Italian Distributor: Tema Ricerca, Bologna, Italy), according to the manufacturer’s instructions. Briefly, following centrifugation, 100 μl of conditioned medium were added in wells precoated with bovine VEGF-A antibody and incubated for 1 h. After washing, the wells were incubated with bovine VEGF-A detection antibody, washed, and then incubated with a solution, provided by the manufacturer, that reacted with the HRP-conjugated antibody. The color reaction was performed using a provided substrate solution, and the absorbance values were measured at 450 nm in a microplate reader (Tecan, Milan, Italy). All the measurements were performed in triplicate. The sample concentrations were calculated from a standard curve and corrected for the number of cultured cells. The results were expressed as ng/ml.
For statistical analysis, the GraphPad Instat statistical package (version 3.05 GraphPad software, San Diego, CA) was used. The data were analyzed by the analysis of variance, followed, when significant, by an appropriate post hoc comparison test (Student's t test if not otherwise indicated in the legends). Differences were considered statistically significant when p values ≤0.05.
We first performed immunocytochemistry experiments on PC and EC in coculture to visualize the architecture of both cell types. Figure 1 shows immunofluorescence images of PC stained with α-actin (Figure 1A) and EC stained with vWF (Figure 1B) antibodies grown on both sides of the filter, and the scheme of preparation of the monoculture (Figure 1C) and coculture (Figure 1D) where PC and EC were grown in contact. We then evaluated the barrier integrity by measuring the TEER and permeability to sodium fluorescein. As reported in Table 1, the TEER of the cell layer was significantly lower in the endothelial monolayer than in the coculture model. Permeability to sodium fluorescein, characterized by low molecular weight, is regarded as a marker for paracellular permeation. Accumulation of sodium fluorescein, diffusing across the endothelial monolayer, was 6.6±0.5×10−6 cm/s, whereas the same parameter was only 3.9±0.21×10−6 cm/s in the coculture, indicating that the contact between EC and PC significantly inhibited the diffusion of this small molecule. These results demonstrate that the contact of pericytes on endothelial cells induces the expression of tight junctions, which reinforce the properties of the blood–retinal barrier. Retinal EC and PC grown on the opposite sides of a porous membrane thus create an in vitro BRB model with reasonably good barrier properties.
To evaluate whether the different culture conditions influence the expression of VEGF, we measured VEGF mRNA and protein levels in the total homogenates of PC and EC from mono- and cocultures. By performing real-time reverse-transcription polymerase chain reaction (RT–PCR) and western blotting analyses, we first noted that, when the basal VEGF content between these two cell types is compared, VEGF expression in terms of mRNA (Figure 2A) and protein (Figure 2B) was higher in PC than in EC. Examining PC separately, we then found that these cells, when in contact with EC, presented decreased intracellular VEGF protein levels compared with monocultured PC (−36%; Figure 2C); conversely, when cultured with PC, EC showed increased intracellular VEGF protein levels compared to EC alone (+100%; Figure 2D). Following the real-time RT–PCR experiments, no changes in VEGF mRNA content depending upon the different culture conditions were observed for PC and EC (not shown). We also evaluated the VEGF protein released in the cell culture medium with ELISA experiments, and we found that, in basal conditions, VEGF levels were higher in the coculture than in the PC and EC monocultures (+198% versus PC; +175% versus EC, respectively; Figure 2E).
We then evaluated the possible variations in PKCβ and HuR protein expression due to the different culture conditions in our cellular models. In particular, we measured PKCβII for PC, and PKCβI and βII in EC, since these latter cells express both β isoforms. Similarly to the intracellular VEGF protein, the PKCβII and HuR basal levels were decreased in the cocultured PC (PCcocu) compared to the monocultured PC (PCmono) (PKCβII: −39%; HuR: −57%, in PCcocu versus PCmono), as reported in Table 2. In contrast, there are no significant differences in the basal PKCβI, PKCβII, and HuR protein content between the monocultured EC (ECmono) and the cocultured EC (ECcocu; Table 2).
We previously found in vitro [16] and in vivo [18] evidence that PKCβ and HuR belong to the same molecular cascade; thus, exploring the effects of PKC activation on the expression of the kinase and, in parallel, on the HuR protein in PC and EC was of interest. First, by performing experiments on other sets of PC and EC, we investigated whether PMA challenge (100 nM PMA for 15 min, the same conditions used in [16]) modified PKCβII levels in these cells. As shown in Figure 3, PKCβII was increased following PMA not only in the PC monocultures (+69%) but also in PC cocultured with EC (+90%).
We then investigated whether PMA stimulation affects total PKCβ protein levels in mono- and cocultured EC. We found the PKCβI and PKCβII isoforms were altered with the phorbol esters treatment; in particular, analogously to PC, the PMA stimulus induced an increase in PKCβII in ECmono and ECcocu (+24% and +91%, respectively) compared to their controls (Figure 3B). Moreover, PMA-treated ECmono presented a trend, although not statistically significant, toward an increase in PKCβI protein content (CTR: 873.9±17.6; PMA: 1107.9±173.6; +27% PMA versus CTR); instead, the upregulation of PKCβI levels was significant in ECcocu following PMA stimulation (CTR: 743.6±38.1; PMA: 1266.1±215.0; +70% PMA versus CTR, p<0.05).
We also evaluated the effects of PKC activation on HuR protein localization and expression in all the cell culture conditions. Immunocytochemistry experiments demonstrated that, following PMA exposure, the HuR protein translocated from the nuclear area to the cytoplasm in PC as well as in EC, in the mono- and cocultures (the latter are shown in Figure 4). Moreover, the PMA stimulus induced an increase in the total HuR protein in PCmono (+44%) and PCcocu (+76%) compared to the relative controls (Figure 3A). Analogously to PC, the HuR protein total content increased in PMA-treated ECmono and ECcocu (+29% and +80%, respectively) versus the control cells (Figure 3B).
Further downstream to the PKC cascade, we first evaluated the possible PMA-induced changes in VEGF expression by measuring the VEGF mRNA and protein content in the mono- and cocultures of PC and EC. In PC, analogously to PKCβII and HuR, PMA exposure was responsible for an increase in the amount of total VEGF protein in both culture conditions. In particular, compared to the relative controls, PMA-treated PCmono and PCcocu presented an 84% and 127% increase, respectively, in VEGF (Figure 3A). Surprisingly, the VEGF mRNA levels in these cells were downregulated by phorbol esters; indeed, the total levels of this transcript decreased (−30%) in PCmono, a decrease that became statistically significant in PCcocu (−68.5%; Figure 5A). Regarding EC, as shown in Figure 3B, following PMA treatment VEGF protein levels increased in ECmono as well as in ECcocu (+91% and +45%, respectively) compared to their controls. Upon PKC activation, VEGF mRNA expression in the EC showed a different profile likely due to the culture conditions: indeed, in the presence of PMA, the VEGF transcript level was higher (+36%) in ECmono, but did not present any significant changes in PMA-exposed ECcocu (Figure 5B).
We then evaluated in all the conditions the effect of PMA on VEGF secretion by performing ELISA experiments on cell culture media. We found no significant variations between the control and the treated cells in the PC and EC monocultures, although a trend to increase was present in the cell types after PMA (not shown). In contrast, in the coculture, in addition to an increase in the intracellular VEGF protein content, PMA also induced a significant increase (44%) in VEGF secretion in the corresponding cell culture medium (COCU: 5.19±0.68 ng/ml; COCU+PMA: 7.46±0.83 ng/ml; p<0.05).
Among the biochemical factors playing a role in the development of retinopathy, such as the ones associated with diabetes, several examples of evidence indicate the importance of the early activation of specific PKC isoforms, mainly βI/βII (reviewed in [8]). Consistently, in vivo studies showed that, in the retina, the production of DAG (or the use of its analogs) induces the preferential activation of PKCβ leading to retinal microvascular abnormalities usually observed in diabetic rats [10,11]. Accordingly, clinical trials have shown that, in diabetic patients, PKCβ selective inhibitors normalize endothelial dysfunction, ameliorate microcirculation, and prevent loss of visual acuity [29]. Of interest, PKC activation is also responsible for the increased expression of VEGF, a key factor in maintaining the normal functions of the retinal vascular cells [12]. As demonstrated by several studies performed with various cell types, PKC-mediated upregulation of VEGF may occur at the transcriptional and post-transcriptional level [8,17,30,31]. We previously suggested that, in retinal pericytes and in the retina itself [16,18], the RNA-binding ELAV/HuR protein may represent a link between PKC and VEGF; in fact, HuR is a PKC target and a post-transcriptional positive regulator of VEGF expression. This latter aspect has been described by others in numerous contexts [32,33]. ELAV proteins primarily act as positive modulators of gene expression, and changes in ELAV-mediated regulation of specific mRNAs have been described in several pathologies [34,35].
To determine the possible mutual influence between PC and EC especially on the expression of VEGF, we cultured PC and EC in direct contact and compared them with the same cells grown separately. Importantly, in our in vitro model, PC and EC were present in the same ratio described in the retina in physiologic conditions, 1:1. Moreover, TEER and permeability analysis (Table 1) indicated that EC when cocultured with PC form a significantly tighter barrier than endothelial cells grown alone. These results demonstrate that our coculture system is a useful in vitro model for studying biochemical alterations occurring in the retinal barrier, thus supporting the use of this system in investigating retinal diseases, including diabetic retinopathy, as reported in the literature [23,36]. We found that in monocultures VEGF mRNA/protein basal levels were more elevated in PC compared to EC (Figure 2A,B), although the amount of the secreted VEGF was comparable between the two cell types (Figure 2E). Moreover, we observed that intracellular VEGF protein content depends upon the different culture conditions; in particular, in the coculture compared to the monocultures, the VEGF protein levels were significantly decreased (−36%) in PC and increased (+100%) in EC (Figure 2C,D, respectively), without changes in the total amount of VEGF mRNA. Interestingly, in spite of these opposite variations in the content of VEGF in PC and EC, the total amount of VEGF protein in the cocultures (PC+EC) remained most likely unchanged compared to the monocultures, suggesting that when PC and EC are cocultured the primary site of VEGF protein synthesis shifts from PC to EC. These data may be read as an adaptive mechanism whereby cocultured PC, to maintain the 1:1 ratio with EC, downregulate their own production of the angiogenic factor VEGF that otherwise may stimulate the proliferation of the cohabiting EC. Of interest, we found that the physical contact between PC and EC produces an increase in the basal secretion of VEGF in the cell culture medium (Figure 2E). Although we cannot determine whether one or both cell types are responsible for this increase, according to the data on intracellular VEGF protein levels, we can hypothesize that EC are the major contributors to this increase in VEGF secretion.
Similarly to VEGF, PKCβII, and HuR protein levels decreased (−39% and −57%, respectively, see Table 2) in the cocultured PC compared to the monocultured PC, again suggesting the importance of keeping the whole PKCβII/HuR/VEGF cascade downregulated in basal conditions. Vice versa, the increased intracellular VEGF protein content observed in the cocultured EC compared to the monocultured EC may be due to the augmented production of this autocrine factor following direct interaction with PC. Accordingly, the influence of PC on the EC protein expression profile has been demonstrated by a recent work illustrating that physical contact of PC upregulates VEGF protein levels in EC [37].
To mimic the consequences of DAG overproduction associated with hyperglycemia, we treated PC and EC with PMA (a DAG analog) and determined the effects of PKC activation on the downstream HuR/VEGF pathway, similarly to a previous in vitro work on pericytes grown alone [16]. In all the examined cultures, PMA-mediated PKC activation led to HuR translocation from the nuclear area to the cytoplasm, and to an increase in the total amount of PKCβ, HuR, and VEGF proteins in the PC and EC. Regarding the underlying mechanisms, there is evidence that the activated PKCβ [38] and HuR [39] enhance their own protein expression. Concerning the increase in VEGF protein levels following PMA, PKC activation likely promotes new VEGF protein synthesis, possibly via the HuR protein, as previously reported [16,18]. Moreover, our ELISA experiments demonstrated that in PMA-treated coculture a parallel increase in VEGF secretion also occurs.
A different profile, perhaps dependent on culture conditions, was observed for VEGF mRNA (Figure 5), whose levels were significantly upregulated by PMA in EC alone, and unchanged in cocultured EC. Concerning PC, the VEGF mRNA levels were not affected by PMA exposure when they were grown in monoculture; conversely, PKC activation led to a significant decrease in the total levels of VEGF mRNA in the cocultured PC. Reading these VEGF mRNA data was difficult and suggested that the changes in the VEGF protein and transcript observed can be ascribed to different and concomitant pathways. This last consideration is in agreement with the fact that VEGF expression can be modulated by other cellular mechanisms besides PKC. However, the finding that, following PMA, VEGF mRNA levels decrease in cocultured PC is in line with the previously mentioned hypothesis that strict control of VEGF production is required for the physiologic coexistence of PC/EC. To this regard, we can also postulate that, in cocultured PC, VEGF protein, whose intracellular levels are increased by PMA, exerts negative feedback on its own transcription [40,41], as described for other growth factors, such as fibroblast growth factor (FGF), whose mRNA and protein can change in the opposite way in response to a specific stimulus [42]. Moreover, considering that PMA also triggers an increased release of VEGF protein in the medium, we cannot exclude a contribution of the secreted VEGF to this negative feedback on PC. In addition, literature data report that in other cellular models VEGF mRNA levels can be downregulated by PMA, although following prolonged exposure [43,44], suggesting that time can be also a decisive and discriminating factor.
In line with our previous in vitro and in vivo works [16,18], we here report new evidence about the pathway involving PKCβ, the mRNA-binding HuR/ELAV protein, and VEGF, which may undergo expression changes triggered by direct PKC activation at early stages of diabetic retinopathy. Moreover, we highlight that PC and EC in coculture show differences in some protein expression profiles, as well as in basal VEGF secretion, compared to the respective monocultures, and that the effects induced by a specific stimulus cannot be predicted by those found in the two cell types grown separately. Therefore, these results suggest that further studies on this in vitro model would be useful to better understand the PC/EC interaction in physiologic and pathological conditions.
This work was supported by a grant to A.P. from the Italian Ministero Università Ricerca (PRIN 2009).