 Molecular Vision
2009; 15:2617-2626
<http://www.molvis.org/molvis/v15/a279>
Molecular Vision
2009; 15:2617-2626
<http://www.molvis.org/molvis/v15/a279> Received 14 October 2009 | Accepted 30 November 2009 | Published 8 December 2009
Molecular Vision
2009; 15:2617-2626
<http://www.molvis.org/molvis/v15/a279>
Received 14 October 2009 | Accepted 30 November 2009 | Published 8
December 2009
Heather L. Grothe,1 Morgan R. Little,1 Angela S. Cho,1 Andrew J.W. Huang,2 Ching Yuan1
The first two authors contributed equally to the work
1Department of Ophthalmology, University of Minnesota, Minneapolis, MN; 2Department of Ophthalmology and Visual Sciences, Washington University, St. Louis, MO
Correspondence to: Ching Yuan, Ph.D., Lions Research Building, Room 326, 2001 6th Street S.E., Minneapolis, MN, 55455; Phone: +02-612-624-9854; FAX: +02-612-625-8880; email: yuanx019@umn.edu
Purpose: Transforming growth factor beta-induced protein (TGFBIp) aggregates into the phenotypic amyloid fibrils and/or non-amyloid deposits in corneal dystrophies and other disorders. While significant progress has been made in molecular genetics to successfully establish the link between the missense mutations of TGFBI and TGFBIp-related corneal dystrophies, the underlying mechanism for the abnormal aggregation remains elusive due to the lack of insights into the conformational perturbations induced by mutations. In the present study, we examined the effects of denaturants and a co-solvent on recombinant TGFBIp, with a focus on protein conformational changes and amyloid fibril formation.
Methods: Recombinant TGFBIp was subjected to various spectroscopic studies, such as far-ultraviolet circular dichroism (far-UV CD), intrinsic tryptophan fluorescence and quenching, and 1-anilinonaphthalene-8-sulfonic acid (ANS) fluorescence, under various denaturing conditions (urea and guanidine hydrochloride [GndHCl], acidic pH, and trifluoroethanol [TFE, co-solvent]). A thioflavin T (ThT) fluorescence assay was used to determine the fibril formation of TGFBIp. In addition, a rabbit polyclonal antibody against the oligomer precursors that initiate the formation of amyloid fibrils was also used in dot blot experiments to detect the formation of prefibrillar precursors.
Results: The purified recombinant TGFBIp is in the folded state according to its intrinsic tryptophan fluorescence analyses. A single-step unfolding process was observed in the GndHCl denaturation experiment. Results from far-UV CD, intrinsic tryptophan fluorescence, and ANS fluorescence experiments showed that TFE exerted its solvent effects by initially unfolding and transforming TGFBIp to a β-sheet-enriched conformer at 20%. When increased to 40%, TFE changed TGFBIp into a non-native α-helix conformer. Although GndHCl and TFE led to protein unfolding, enhanced fibril formation could only be observed in the presence of TFE and at acidic pH, according to the ThT fluorescence assays. The paradigmatic protofibrillar TGFBIp oligomers were also detected during the fibril formation by the dot blot experiment.
Conclusions: Our results suggest that protein unfolding may serve as the prerequisite but is not sufficient for the fibrillogenesis. Other factors, such as the solvent used, fragmentation, or pH, may also be crucial for the formation of TGFBIp fibrils.
Pathological protein aggregations, particularly amyloidosis, affect almost all tissues and organs and have been implicated in various human diseases, such as Alzheimer’s disease, Creutzfeldt-Jakob disease, familial amyloidotic polyneuropathy, Parkinson’s disease, and others [1-3]. Corneal amyloidosis disorders, including gelsolin-related lattice corneal dystrophy type II (Finnish-type familial amyloid polyneuropathy type IV), lactoferrin-related familial subepithelial amyloidosis, transforming growth factor beta-induced gene (TGFBI)-related corneal dystrophies, and polymorphic amyloid degeneration, are characterized by phenotypic abnormal deposits accompanied by corneal opacities, epithelial abnormalities, pain, and potential loss of vision [4]. In addition to those pathogenic gene mutations that either have systemic effects or specifically affect only corneal tissues, secondary amyloidosis has been reported in various ocular disorders, such as keratitis, chronic post-traumatic inflammation, glaucoma, keratoconus, trichiasis, and even Fuch’s endothelial dystrophies [5-7]. Among these various types of corneal amyloidosis disorders, TGFBIp-related corneal dystrophies are perhaps the most well studied.
TGFBIp (the protein product of the TGFBI gene, also referred to as keratoepithelin, BIGH3, beta-igh3, βigh3, or RGD-CAP) is a 683-amino acid secretory matrix protein induced by transforming growth factor β-1. It plays essential roles in cell adhesion, differentiation, and tumorigenesis [8-13]. The temporal expression of TGFBI during embryogenesis and wound healing suggests its important roles in the development and maintenance of ocular surface integrity [14-16]. Primary sequence analysis reveals that the encoded TGFBIp contains a secretory signal peptide sequence in the NH2-terminus, and an arginine-glycine-aspartate (RGD) motif located at the COOH-terminal end, which can mediate cell adhesion and other functions via integrins [17]. In addition, there are four homologous repeats designated as “Fas-like” domains in TGFBIp [18]. The RGD motif in TGFBIp may not be involved in integrin-mediated cell adhesion, but rather in other events, such as apoptosis [19,20]. Mass spectroscopy has indicated that mature TGFBIp from CHO cells does not contain the RGD motif, likely as a consequence of post-translational degradation [21]. Instead, novel motifs (NKDIL and EPDIM) located in the second and fourth Fas-like domains were identified as exerting an integrin-binding capability [10].
The pathogenic roles of mutant TGFBIp in certain types of stromal corneal dystrophies have drawn great attention since TGFBIp’s discovery in 1992 [8]. Genetic analyses [22] have linked several mutations of the TGFBI gene to various corneal dystrophies, including Avellino, lattice dystrophies, Reis-Bucklers dystrophies, and granular dystrophies. Currently, more than 30 missense mutations of TGFBI have been identified and linked to at least 13 different phenotypes of corneal dystrophies, characterized by the presence of abnormal amyloid fibril and/or non-amyloid deposits in sub-epithelial and stromal layers in the cornea [4,23,24]. Painful corneal erosions often recur, likely due to poor epithelial adhesions. Interestingly, almost all the dystrophy-related mutations are clustered in the first and fourth Fas-like domains (Fas-1 and Fas-4). While the role of genetic predisposition has been well established, it is likely that the genetic mutation is not the only determinant contributing to the amyloid fibril and non-amyloid aggregations in TGFBIp-related corneal dystrophies. The fact that both wild-type and mutant recombinant TGFBIp readily form amyloid fibrils [10] strongly suggests an intrinsic propensity of TGFBIp toward aggregations and a potential pathogenesis mechanism. Previous studies also indicate that TGFBIp contains amyloidogenic motifs that play essential roles in the aggregation mechanism, similar to other disease-related amyloidogenic proteins such as amyloid beta precursor protein or lysozymes. Using synthetic peptide approaches, at least two endogenous amyloidogenic motifs on TGFBIp, spanning from residues 110 to 131 and from 515 to 532, respectively, have been identified [25-27]. However, the role of these amyloidogenic motifs for amyloid fibril formation within the full-length protein context awaits further investigation. In addition, tissue-specific protein or matrix degradation may also play roles in the pathogenesis of the abnormal aggregations of TGFBIp [28].
Protein conformational change and unfolding are proposed to be the prevailing mechanisms for amyloid fibril formation [29]. Studying the molecular properties of disease-related proteins under amyloid-conducive conditions should shed light on the protein aggregation behavior and related pathogenic pathways. Furthermore, studies with recombinant proteins are currently the only avenue for understanding the mechanism of amyloid fibril formation of TGFBIp, since transgenic animal models have failed to manifest corneal dystrophic phenotypes [30]. In this study, we set forth to characterize the conformational properties of TGFBIp with spectroscopic tools, and have investigated the denaturation profile and the effects of pH and solvent on its conformations and fibril formation.
All chemical reagents were purchased from Sigma Chemical Co. (St. Louis, MO), if not otherwise specified. Puromycin was purchased from Clontech (Mountain View, CA). Rabbit anti-oligomer polyclonal antibody that recognizes the amino acid sequence-independent oligomeric conformers of various amyloidogenic proteins was purchased from BioSource International (Camarillo, CA). Rabbit anti-TGFBIp polyclonal antibody was custom-made by Bethyl Laboratory (Montgomery, TX). E. coli-expressed full-length recombinant TGFBIp was used as the antigen for immunization [31].
The TGFBIp coding sequence from 1 to 641 residues with the RGD motif removed (resembling the matured form of the secreted protein) was PCR-amplified from an I.M.A.G.E. clone (Clone I.D. 2957915; Genbank BE206112), and inserted into pIRES.puro3 for stable expression in mammalian cells (293FT; Invitrogen, Carlsbad, CA). (His)6 and Strep II tags were engineered to be expressed right after the encoded signal peptide and in the COOH-terminal end, respectively, to facilitate the subsequent purification. After transfection, cells were maintained in Dulbecco's Modified Eagle Medium plus 10% fetal bovine serum under puromycin selection (3 μg/ml) for two weeks. Individual clones were selected according to their growth rates and TGFBIp expression level (as determined by dot blot experiments). The TGFBIp-expressing clones were further expanded in a serum-free medium system (FreeStyleTM 293, Invitrogen) with reduced puromycin (1 μg/ml). Secreted TGFBIp in the conditioned medium was loaded onto a Ni+-NTA column for single-step purification. The purities of recombinant WT TGFBIp were confirmed by SDS-PAGE gels and were routinely higher than 95%. Purified TGFBIp were further concentrated using Centriprep YM-30 (Amicon, Bedford, MA) to the intended concentrations determined by the BCA assay (Pierce, Rockford, IL).
Fluorescence studies were performed using a FluoroMax-II spectrofluorometer (Jobin Yvon-SPEX, Edison, NJ). Intrinsic tryptophan fluorescence spectra of recombinant TGFBIp (approximately 0.1 mg/ml in 50 mM Tris-HCl, 150 mM NaCl, pH 7.4) were taken in a 1 cm quartz cuvette at 25 oC. Quenching experiments were carried out by adding aliquots of acrylamide or potassium iodide (KI) stock solutions (5 M) sequentially into recombinant TGFBIp samples. The excitation wavelength was 295 nm (tryptophan) and the emission spectra were scanned from 300 to 500 nm. Sodium thiosulfate (0.1 mM) was added to the KI stock solution to prevent the formation of I3-, which has a yellowish color that interferes with the spectroscopic measurement. The emission intensities at 330 nm in the absence and in the presence of a quencher (Fo and FOBS) were used for the Stern-Volmer analyses [32,33]. The data were further corrected for the dilution and the inner filtering effect accordingly [34,35]:
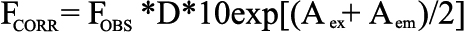 ,
,where FCORR and FOBS are the corrected and observed fluorescence, respectively, and D is the dilution factor due to the volume increase from the added quencher; Aex and Aem are the absorption of the quencher at the excitation and emission wavelengths, respectively. As the plot of F0/F versus acrylamide appeared to be linear (see Results), the two tryptophan residues (TGFBIp contains two tryptophan residues, W68 and W148) likely have heterogeneous fluorescence behaviors, but only differ by a factor of less than 2 within the experimental error [32]. Therefore, the effective quenching constant, KSV(eff), was determined by plotting (Fo/F-1)/[Q] versus [Q] ([Q], concentration of the quencher) and extrapolating to [Q]=0, where the initial slope can be obtained for the value of KSV(eff).
For the denaturation experiments, the emission maximum (“Emax”) and intensity-averaged emission maximum (“IAEM,” [λ]=Σ (λi*Ii) / Σ λi, where Ii is the fluorescence intensity at the individual wavelength λI) of tryptophan residues were used as indicators for the unfolding/denaturation of TGFBIp in the presence of guanidine hydrochloride (GndHCl) or urea [36]. For Thioflavin T (ThT) assays, 100 μl samples were mixed with 700 μl of 25 μM ThT in 50 mM glycine-NaOH, pH 8.5, and then immediately measured for their ThT fluorescence spectra (excited at 450 nm and scanned from 460 nm to 600 nm). For the pH-dependent amyloid fibril formation, recombinant TGFBIp were mixed with the buffering solution containing 150 mM NaCl and 50 mM of sodium citrate, MES, MOPS, Tris, CHES, and CAPS [27], with pH ranging from 2 to 13. The formed fibrils were further spun down by a TL-100 ultracentrifuge (Beckman Coulter, Fullerton, CA) at 38,500× g (25,000 rpm), washed once in 50 mM Tris-HCl, 150 mM NaCl, pH 7.4 solution and then resuspended in ThT solution for the ThT fluorescence assays to reduce the interference by pH. The fluorescence of 1 anilino-naphthalene-8-sulfonic acid (ANS) was used as a spectroscopic tool to investigate the hydrophobicity of recombinant TGFBIp in various conditions. Samples in 50 μM ANS, 50 mM Tris-HCl, pH 7.4 were measured using an excitation wavelength of 350 nm and scanned from 400 to 600 nm.
The far-UV circular dichroism (CD) spectra were measured by a Jasco J-710 spectropolarimeter (Japan Spectroscopic Co., Tokyo, Japan). Each spectrum was obtained from the average of ten scans with blank-subtraction, smoothed by a binomial smoothing routine, and plotted using Kaleidagraph software (Synergy Software, Reading, PA).
Samples of TGFBIp were incubated at 37 oC in 50 mM Tris-HCl, 150 mM NaCl, pH 7.4 for up to one week. Five microliters of sample were applied onto a carbon-coated formvar copper grid and stained with 2% phosphotungstic acid at pH 7.4 at room temperature for 2 min. The fibrils were examined with a JEOL 1200 transmission electron microscope at the University of Minnesota Characterization facility.
In order to obtain highly pure, homogeneous TGFBIp for biochemical and biophysical studies, we have constructed and evaluated various expression vectors (Figure 1A) for recombinant protein production. In contrast to the extensive degradation patterns of recombinant TGFBIp (lane 1, Figure 1B) produced by the single-tagged construct (“[His]6-TGFBIp”, Figure 1A), the double-tagged construct with the RGD motif removed (“[His]6-TGFBIp-[Strep II]”, Figure 1A) generates a single major protein product (lane 2, Figure 1B). On average, 6–8 mg of purified recombinant TGFBIp was obtained from 1 liter of conditioned medium using a single-step Ni+-NTA affinity chromatography, with purities greater than 95%, as demonstrated by SDS-PAGE gel electrophoresis. Previously, WT TGFBIp and peptides have been shown to form fibrils in vitro by various researchers [25-27,37]. Recombinant TGFBIp produced in this study formed fibrils, as demonstrated by the ThT fluorescence assay (Figure 1C) and electron microscopy (Figure 1D) when incubated in the Tris-buffered saline (1× TBS) at 37 oC for one week. Fibrils of 5–10 nm in width and up to several hundred nm in length were observed after extensive incubation at 37 oC (Figure 1D).
The intrinsic tryptophan fluorescence of recombinant WT TGFBIp was studied for the denaturation profile using urea and GndHCl. The tryptophan residues display a fluorescence emission peak at 330 nm (Ex=295 nm) in the native state (Figure 2A, 0.0 M GndHCl), indicating that they are in a relative hydrophobic environment. Results from quenching experiments revealed that acrylamide, a non-ionic polar quencher, is able to quench the intrinsic tryptophan fluorescence effectively (Figure 2A, inset). I-, on the other hand, failed to produce evident quenching below the concentration of 1 M. Stern-Volmer analyses of the quenching experiments by acrylamide revealed a linear plot, suggesting that the collisional constants of the two tryptophans differ by less than two-fold [32]. The effective collisional quenching constant KSV(eff) was determined to be 4.63 M-1.
Exposure of tryptophan to the hydrophilic environment during protein unfolding results in a redshift of the emission maximum and a change in the quantum yield. The addition of 1.0 to 6.0 M GndHCl produced a significant redshift of the emission maximum from 330 to 355 nm, along with a significant reduction of fluorescence intensity, as shown in Figure 2A. The concentration of the half-transition by GndHCl denaturation was determined to be approximately 3.5 M (Figure 2B), by both Emax and IAEM. The latter index was calculated from the entire spectrum and reflects both the shape and position changes, and is therefore less susceptible to error [36]. The curve-fittings with either indexe also suggested a simple two-state denaturation profile (native and denatured states) by GndHCl. Reduction of the total fluorescence intensity was also observed with increasing GndHCl concentration that approximates a two-state transition (data not shown). Urea at concentrations of up to 8 M, on the other hand, failed to denature recombinant TGFBIp (Figure 2B, inset) and merely produced a 4 nm shift of the emission maximum (from 330 to 334 nm), in contrast to the 25 nm redshift caused by 6 M GndHCl. When WT TGFBIp was incubated with GndHCl at various concentrations that either partially (1 or 2 M) or fully (4 or 6 M) denatured the protein, no fibril formation was observed according to the ThT fluorescence assays (Figure 2C).
The pH effects on the fibril formation of TGFBIps were investigated by incubating recombinant proteins at various pHs for 24 h at 37 oC. TGFBIps were instantly denatured in alkaline pH (pH 13), as demonstrated by the prominent redshift of the emission maxima of the tryptophan residues (“pH 13”, Figure 3A inset), while aggregations of TGFBIp were promoted in acidic pHs conditions, judging from the elevated light scattering from the baseline (“pH 3” versus “pH 7” in Figure 3A, inset). Significant increases of fibril formations measured by ThT fluorescence assay were noted at conditions below pH 6 (Figure 3A) and reached a plateau at pH 3. On the other hand, the ThT fluorescence intensities remained minimal above pH 6. SDS-PAGE analyses showed that increased fragmentation of TGFBIp occurred at pH 3 after 24 h incubation (37 oC, Figure 3B). Such a degradation in acidic pH was further enhanced by elevating the incubation temperature to 60 oC (Figure 3B).
As solvation plays essential roles in amyloid fibril formation, we further investigated the solvent effects of trifluoroethanol (TFE) on the conformation and fibril formation of recombinant TGFBIp. The emission maxima of the intrinsic tryptophan fluorescence of TGFBIp were approximately 330 nm in 0–10% TFE (Figure 4A), with the slight reduction of the intensities likely due to the reduced quantum yield in the presence of TFE. At 20% TFE, significant conformational changes of TGFBIp occurred, which is indicated by the shift of the emission maximum toward the red (approximately 348 nm), along with some degree of aggregations reflected by the uprising of the baseline toward the blue (light scattering). At 40% TFE, the tryptophan residues further redshift to approximately 351 nm.
ANS fluoresces to a much higher degree when it binds to exposed hydrophobic patches, and therefore has been used as an indicator for protein unfolding. When ANS was used to probe the unfolding of TGFBIp at various TFE concentrations, minor changes of ANS fluorescence were noted from 0 to 10% TFE (Figure 4B). A prominent ANS increase was noted at 20% TFE, indicating that a conformational change of TGFBIp occurred. At 40% TFE, the ANS fluorescence was significantly reduced.
The far-UV CD spectroscopy (Figure 4C) showed that at sub-denaturation TFE concentrations (0 to 10%), TGFBIp appeared to be in a predominantly α-helix conformation. At 20% TFE, a global conformational change of TGFBIp took place as the α-helix was reduced, with an instant increase of the ellipticity at 218 nm to β-sheet-dominant species. At 40% TFE where the co-solvent forms dynamic clusters, TGFBIp was converted to a non-native α-helix conformer, judging from the prominent 208/222 nm troughs (Figure 4C).
The solvent effects on the fibril formation were studied by incubating TGFBIp with TFE at 37 oC for 24 h and then measured for its ThT fluorescence. The conformation of TGFBIp at 0 or 5% TFE remained unchanged after extensive incubation (48 h, data not shown) and did not produce fibrils in vitro, whereas significant fibril formation was noted in 10 to 20% TFE, with a slight reduction at 40% TFE after 24 h incubation (Figure 4D). Increasing the TFE concentration seemed to affect both the yields and the kinetics of fibril formation (Figure 4D, inset). The fibril formation for TGFBIp at 20% TFE had a fast onset and produced the highest signal at 4 h with a slight decrease subsequently. The 40% TFE produced a quick formation of fibril initially, which was also supported by the results of oligomeric protofibrillar formation (cf., Figure 4E, inset), but the ThT signal remained unchanged subsequently. TGFBIp at 10% TFE showed a slow fibril formation, but reached a level higher than that at 40% TFE.
While 10% TFE did not unfold TGFBIp initially, it eventually induced conformational changes after extended incubation, as indicated by the CD spec (Figure 4E). Using a polyclonal rabbit antibody that specifically recognizes the universal oligomeric conformers of fibrillar precursors [37], we further demonstrated that TGFBIp formed oligomers in the presence of 10% TFE after 2 h, which could be seen in the dot blot assays used to monitor the oligomeric protofibrillar formation (Figure 4E, inset). On the other hand, no formation of oligomer could be detected at 0 or 5% TFE. Prominent oligomer formations were detected at higher concentrations (20 and 40%) starting 0.5 hour after incubation with TFE. The blots were also probed with the α-TGFBIp antibody to ensure equal loading of proteins in individual wells (data not shown).
Extensive and variable post-translational degradation patterns have been observed for TGFBIp either extracted from tissues, secreted by cultured cells or produced from ectopic mammalian expression systems [19,21,28,38,39], with or without the RGD motif. A comparison study of TGFBIp fragments extracted from normal and diseased corneas (R124C, R124H, and R124L) by 2-D gel electrophoresis further demonstrated distinct degenerative degradations in both the N- and C-termini, suggesting differential proteolysis of mutant proteins [28]. The current construct (“[His]6-TGFBIp-[Strep II]”) used in our study has generated a highly purified single TGFBIp protein, compared to the multiple protein products expressed from the (His)6-TGFBIp construct or extracted from tissues. The homogeneity and ease of purification of this slightly truncated, matured TGFBIp circumvent the difficulties in obtaining TGFBIp samples with high purity and bioactivity (such as the cell adhesion assay) for biochemical and biophysical experiments. As demonstrated in Figure 1C,D, the purified recombinant TGFBIp can also form amyloid fibrils similar to what other researchers have previously reported [20].
The recombinant TGFBIp displays a two-state denaturation profile by GndHCl, but not by urea. As urea is a nonionic chaotropic agent that denatures proteins by disrupting the hydrophobic interaction, it is likely that additional factors such as electrostatic interaction play significant roles in the folding of TGFBIp. GndHCl, on the other hand, is an ionic chaotropic species that disrupts both hydrophobic and electrostatic interactions, which can fully denature TGFBIp, judging from the emission-maximum shift of the intrinsic fluorescence. TGFBIp contains a cystine-rich domain in the NH2-terminus, which potentially can contribute to the folding of TGFBIp via the intramolecular covalent disulfide bond; however, incubating TGFBIp in the presence of urea and reducing agent (dithiothreitol) did not lead to further denaturation (data not shown), which does not support the role of disulfide bond in the folding of TGFBIp. The insights gained from our GndHCl denaturation study will be useful for a future comparison study of dystrophy-causing mutants and WT TGFBIp.
Protein conformational changes and unfolding induced by mutations, oxidation, and other factors have been proposed to be the pathogenesis mechanism for amyloid fibril aggregations [29,40]. One hypothesis for the abnormal aggregation of amyloidogenic proteins is that conformational change and/or protein unfolding exposes the endogenous amyloidogenic regions and facilitates their aggregations into prefibrillar oligomer precursors, fibrils, and eventually amyloid deposits [41]. It has been shown previously that denaturation or partial unfolding by GndHCl can promote amyloid fibril formation in various proteins. For example, GroES (an hsp10 homolog from E. coli) readily forms fibrils when partially denatured by GndHCl [42]. However, incubating TGFBIp at partially (1 and 2 M) or fully denaturing (4 M) concentrations did not lead to fibril formation (Figure 2C). Similarly, human and rat hsip10 with sequence homology to GroES failed to form fibrils under GndHCl-perturbed states in a recent study [43]. It is likely that while GndHCl denatures these proteins and exposes their endogenous amyloidogenic regions, it may also disrupt the intermolecular interactions essential for cross-β oligomerization.
Acidic pH conditions promote fibril formation of Aβ peptides, prions, stefin B [44-46], and some non-disease amyloidogenic proteins, as well as TGFBIp (Figure 3A). In addition to the destabilizing effect on protein conformation, low pH conditions can also accelerate the chemical cleavage of peptide bonds of aspartate and glutamate residues [47], which can lead to peptide fragmentation. Such an effect on peptide fragmentation can be further enhanced by high temperatures [48,49]. We observed similar fragmentations and temperature effects with recombinant TGFBIp incubated under low pH conditions (Figure 3B). While extreme pH is rarely encountered under normal physiological conditions (except in the lysozyme), fragmentation of proteins is a key factor in initiating fibril formation in vivo. Peptide fragmentation has been shown to trigger amyloid fibril aggregations of lactoferrin, amyloid-β protein, gelsolin, and other proteins [41,50,51]. Deposits of differentially degraded TGFBIp fragments were noted in the diseased corneas from dystrophic patients, and may likely contribute to the pathogenesis of TGFBIp-related corneal dystrophies [28]. Further study on TGFBIp fragments generated by limited proteolysis or expressed from recombinant sources will help to shed light on the molecular mechanism of TGFBIp amyloidosis.
TFE has been used widely to investigate the solvent effects on amyloidogenic peptides and proteins. With a low dielectric constant, and being mildly more acidic than water, TFE promotes the unfolding and increases the intramolecular hydrogen bonds in proteins [52]. At low concentrations, TFE destabilizes the specific tertiary interactions of native proteins; at higher concentrations, it has been known to stabilize secondary structures (the α-helix) and induce non-native folded states of proteins [53,54]. TFE can also form micelle-like dynamic clusters at higher concentrations (>30%) and modulate the molecular properties of proteins. At the denaturing concentrations, TFE often promotes significant β-sheet formation, and subsequently leads to fibril formation [55]. These effects of TFE on the protein conformation of TGFBIp were also observed. It is clear that TGFBIp formed the non-native α-helical conformers at a high TFE percentage (40%) from CD and intrinsic fluorescence results (Figure 4A,C). At 20% TFE, TGFBIp immediately transformed from an α-helix-enriched native state to a partially denatured state containing predominantly β-sheets (Figure 4C). The transformation not only promoted the aggregating of TGFBIp into the cytotoxic oligomer (a fibrillar precursor, Figure 4E), but also eventually led to prominent fibril formation, according to the ThT fluorescence assays (Figure 4D). Although initially TGFBIp remained in its native state at a sub-denaturation concentration (10%), extended incubation (after 2 h) gradually unfolded TGFBIp and produced oligomers and amyloid fibrils (Figure 4E).
In summary, our study has provided spectroscopic characterizations of recombinant TGFBIp in the presence of denaturants and a co-solvent. Protein unfolding may not be the only determinant for the fibril formation of TGFBIp. Other factors, such as solvents, fragmentation, and pH conditions, also play critical roles. Comparison studies of the molecular properties of WT and dystrophic-related mutant proteins (such as R124C for lattice type I) are currently underway in order to shed light on the pathogenesis mechanism of TGFBIp-related corneal dystrophies.
This study was supported in part by grants from: Eye Bank of America Association, Research to Prevent Blindness, Minnesota Medical Foundation, Fight For Sight Inc. and National Eye Institute (EY017609 to A.J.W.H. and EY017852 to C.Y.). We would like to thank Dr. Deborah Ferrington for her critiques and suggestions on the manuscript.