Figure 2. In silico structure analysis of the missense ABCA4 variants. The variant models (blue) are superimposed and compared with the wild-type (WT) model or experimental structures
(gray). The WT residues are shown as yellow sticks, and substitutions are shown as purple. The red discs show van der Waals
(vdW) overlaps or steric clashing with the substitution. They are given only when every possible side-chain conformation resulted
in clashes in experimental and predicted structures, but only the highest possibility conformation is shown here. A. C54Y
results in disulfide bond breakage and clashing interactions. B. R212C leads to an intra-domain H-bond breakage. C. L541P
disrupts an α-helix and produces steric clashes. D. A1038V causes slight conformational change. E. R1098C causes the loss
of a salt bridge affecting NBD1–RD2 domain–domain interaction; the RD2 is colored light green. F. T1253M inside the RD1 leads
to a loss of the H-bond and a premature β-sheet. G. P1380L results in clashing interactions with neighboring residues as well
as changes in the swivel angle of the transmembrane helix and indirectly breaks H-bonds with His-1365 and phosphatidylethanolamine
(PE; shown in pink). This proline-induced kink in the WT transmembrane domain may have a functional role in substrate transport
across the membrane. The in silico variant (blue) model is superposed onto the cryo-EM structure of the human ABCA4: 7e7o
(gray) to show the distortion. H. A1598D results in steric clashes and introduces a buried hydrophilic residue with an expected
destabilizing effect. I. G1961E causes clashing interactions and replaces a buried hydrophobic residue with a hydrophilic
amino acid. J. The E2233V novel variant in the RD2 results in H-bond breakage with a residue in the NBD1 domain (light green).
Visualized and analyzed in PyMOL2.
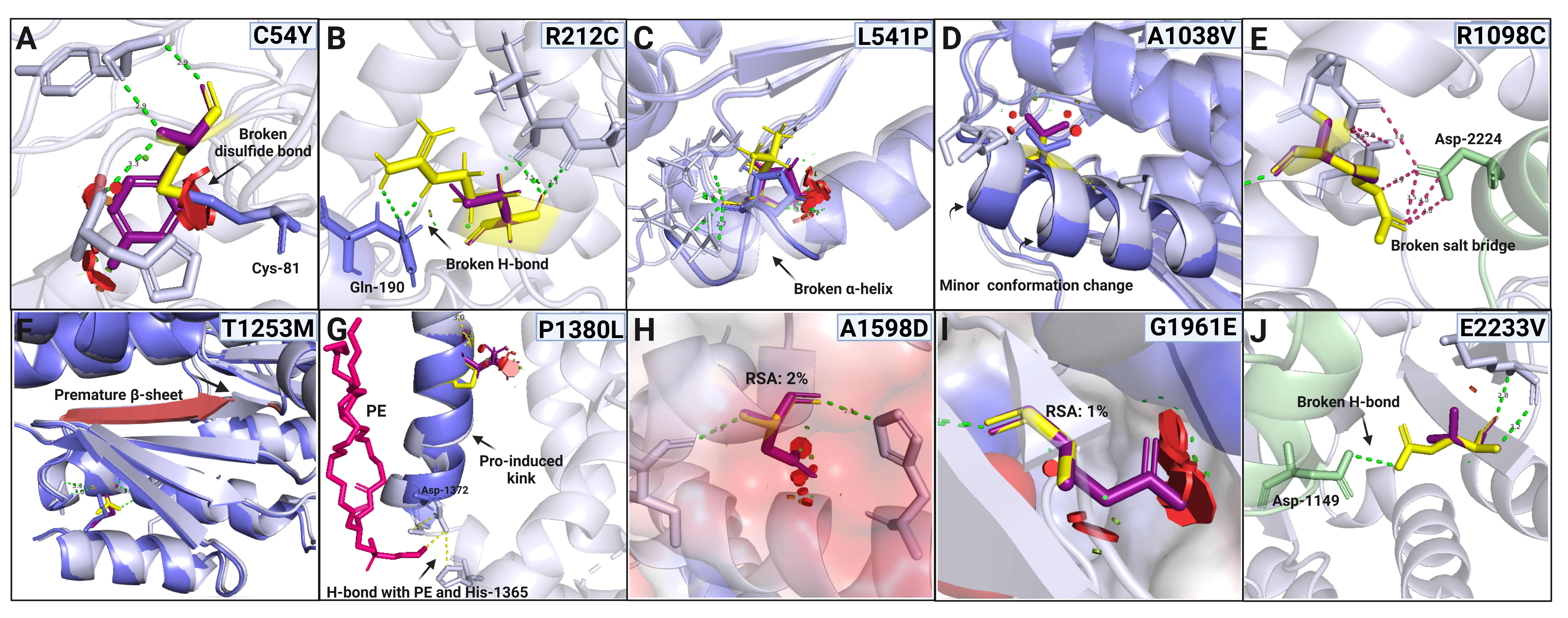
 Figure 2 of
Cevik, Mol Vis 2023; 29:217-233.
Figure 2 of
Cevik, Mol Vis 2023; 29:217-233.