Figure 2. Multiple sequences alignment of CYP1B1 orthologs and molecular modeling of missense variants. A: ClustalW alignment of CYP1B1 proteins shows conservation of the residues at positions 181, 290, 390, 404, 437, 442, and
479 among eight species. B: Predicted three-dimensional (3D) structures of wild-type and mutant CYP1B1 proteins created using Phyre2. The positions
of the wild-type and mutated forms of the amino acids in this cohort are shown in green and red, respectively. The p.Leu181
residue of CYP1B1 is located in the α-helix of the secondary structure, and replacing this residue with a smaller, less hydrophobic
glutamine residue is predicted to slightly destabilize the local conformation and leads to the loss of hydrophobic interactions
in the protein core. Modeling of the cytochrome domain of CYP1B1 revealed that the p.Arg390 residue forms a hydrogen bond
and a salt bridge with glutamic acid at position 387, asparagine at position 428, and proline at position 437. Substitution
with smaller residue histidine [p.(Arg390His)] or cysteine [p.(Arg390Cys)] is predicted to disrupt this hydrogen bond, and
interrupt the signal transduction between the two domains of CYP1B1, cause an empty space in the core of the protein, and
distort the correct protein folding. The p.Pro437 residue is located on the protein’s surface. Prolines are known to be rigid
and therefore, induce a special backbone conformation. Replacing a proline with leucine might disturb this special conformation
and distort the interactions with other molecules. Finally, the substituting glutamine with arginine at position 479 is predicted
to affect the protein stability by introducing protein-folding problems due to its positive charge, which, in turn, could
affect the ligand contacts made by one of the neighboring residues.
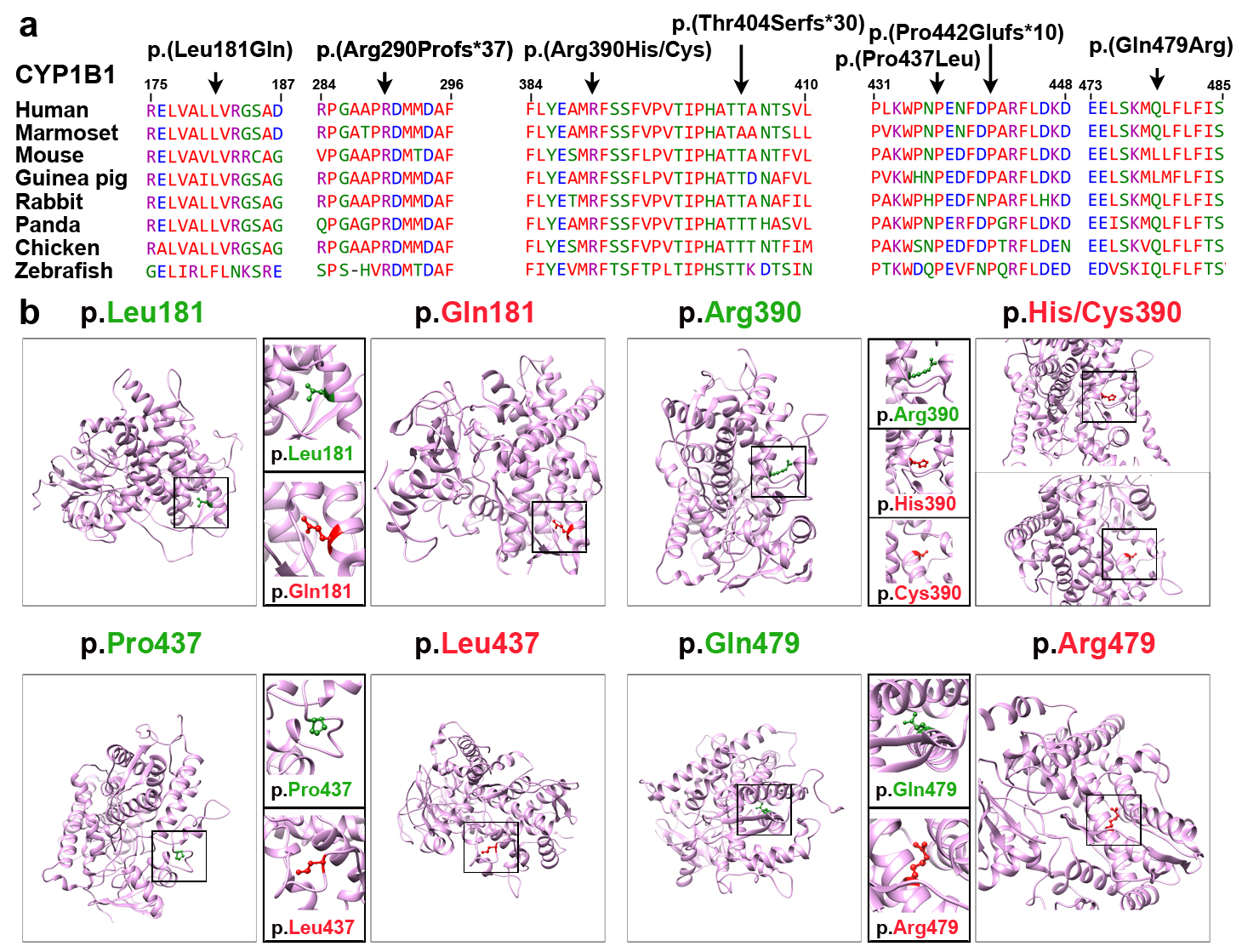
 Figure 2 of
Rashid, Mol Vis 2019; 25:144-154.
Figure 2 of
Rashid, Mol Vis 2019; 25:144-154.