Figure 1. The basis and pipeline of circRNAs identification and analysis. A: After high throughput sequencing, raw data were collected and subsequently filtered and analyzed for quality control (QC).
Clean data were mapped on the genome, and then the circular RNAs (circRNAs) were predicted via CIRI (CircRNA Identifier) and
the circBase database. Finally, analyses of the differential expression between the time points, as well as function and origin
analyses, were performed. B: Two segments of junction reads of classic circRNAs match to the relevant sequence in reverse orientation, albeit separately.
C: If a flanking part of the junction is a short exon, the segment can align to the flanking exon of the circRNAs. D: If the circRNAs length is short, the read will possibly contain two terminal segments that align to the area of junction
within circRNA. E: To reduce the false positive rate, candidate circRNAs are filtered via criteria as in a previous study.
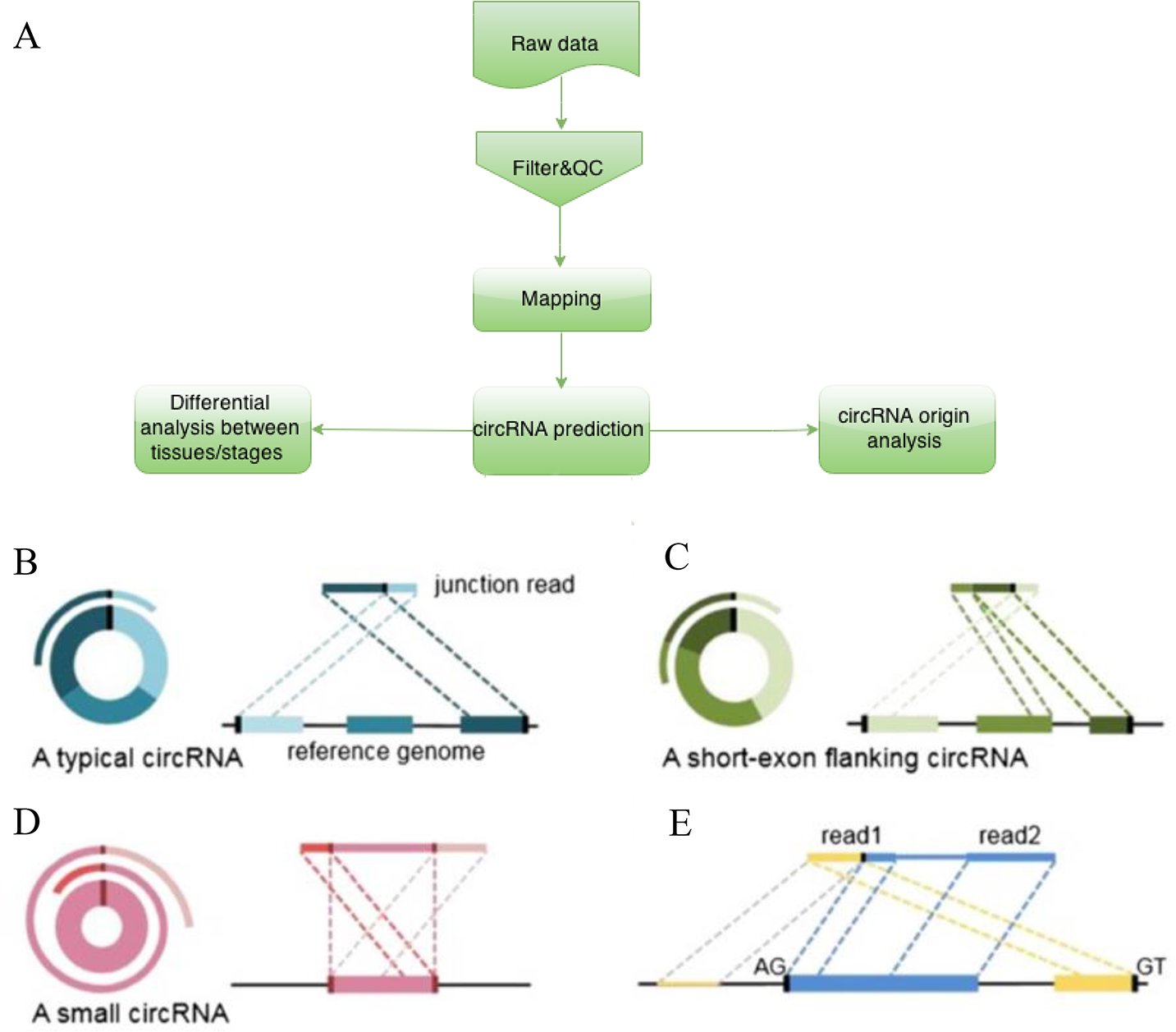
 Figure 1 of
Han, Mol Vis 2017; 23:457-469.
Figure 1 of
Han, Mol Vis 2017; 23:457-469.