ABSTRACT
Purpose: Mutations in the retinal degeneration slow (RDS)/peripherin gene have been shown to be associated with pattern dystrophy of the retina (PDR) and other retinal dystrophies. The aim of our study was to confirm or exclude the RDS locus and the rhodopsin (RHO) locus as the disease causing locus in a large Swiss family affected with pattern dystrophy of the retina.
Materials and Methods: A Swiss family with 14 members across 3 generations affected with PDR was examined. Eleven living family members were investigated using 6 markers surrounding the RDS and RHO loci.
Results: Linkage to two possible candidate genes, the RDS gene on chromosome 6p and the rhodopsin gene on chromosome 3q, could be excluded.
Conclusions: The family provides evidence for genetic heterogeneity of PDR and is in agreement with heterogeneity in other retinal dystrophies. Further investigations are in progress to map the gene causing PDR in this family.
INTRODUCTION
Pattern dystrophy of the retina (PDR) represents a clinically defined subgroup within photoreceptor dystrophies. Lesions predominate in the central fundus and exhibit considerable phenotypic heterogeneity. Different phenotypes have been observed in the same family (1, 2, 3) and age-dependent expression of the different alterations has been described (4). Common characteristics of PDR include autosomal dominant inheritance, electrophysiological changes (manifested mainly in the electrooculogram), and a fair to good visual prognosis. Fluorescein angiography is a sensitive tool for the confirmation of PDR.
Recently, mutations in the retinal degeneration slow (RDS)/peripherin gene on chromosome 6p were found to be associated with retinitis pigmentosa, macular dystrophies, fundus flavimaculatus, retinitis punctata albescens and pattern dystrophy of the retinal pigment epithelium (5, 6, 7, 8, 9). The RDS gene is expressed in rods and cones of the retina, but its function is still unknown (10). Interestingly, in one family the same mutation resulted in different phenotypes including retinitis pigmentosa, pattern dystrophy and fundus flavimaculatus (11).
Mutations in the rhodopsin (RHO) gene were found in about 25% of patients with autosomal dominant retinitis pigmentosa and in an unknown percentage having congenital stationary night blindness (9). More than 60 different mutations in this gene have been described. Although rhodopsin is only expressed in rods, mutations in this photoreceptor protein affect both rods and cones (12). The RHO gene has to be considered as another possible candidate gene/locus for PDR.
With the rapidly growing knowledge of genes responsible for inherited eye disorders, genetic heterogeneity has been documented for several photoreceptor dystrophies (13), but not for PDR. As part of an ongoing study, a large Swiss familiy affected with autosomal dominant PDR was investigated (4). The aim of the present study was to exclude or confirm the involvement of RDS or RHO in the pathogenesis of PDR in this family. Additionally, studying retinal PDR may contribute to a better understanding of age related macular degeneration.
MATERIAL AND METHODS
Figure 1 shows a part of the pedigree of a large Swiss family with more than 80 living members. All family members who participated in the study (n=64) underwent a thorough examination including medical and ophthalmic history, measurements of best corrected visual acuity and intraocular pressure, slit-lamp, and fundus examinations. Patients with suspicious findings were further investigated by fluorescein angiography. Ages of the patients at the time of clinical examination ranged from 32 to 67 years. The male/female ratio of affected family members was 4:3. Pattern dystrophy could be confirmed in 13 family members. Eight of these patients, aged 32 to 67 years, had no symptoms (4).
Figure 1
Pedigree of the Swiss PDR family.
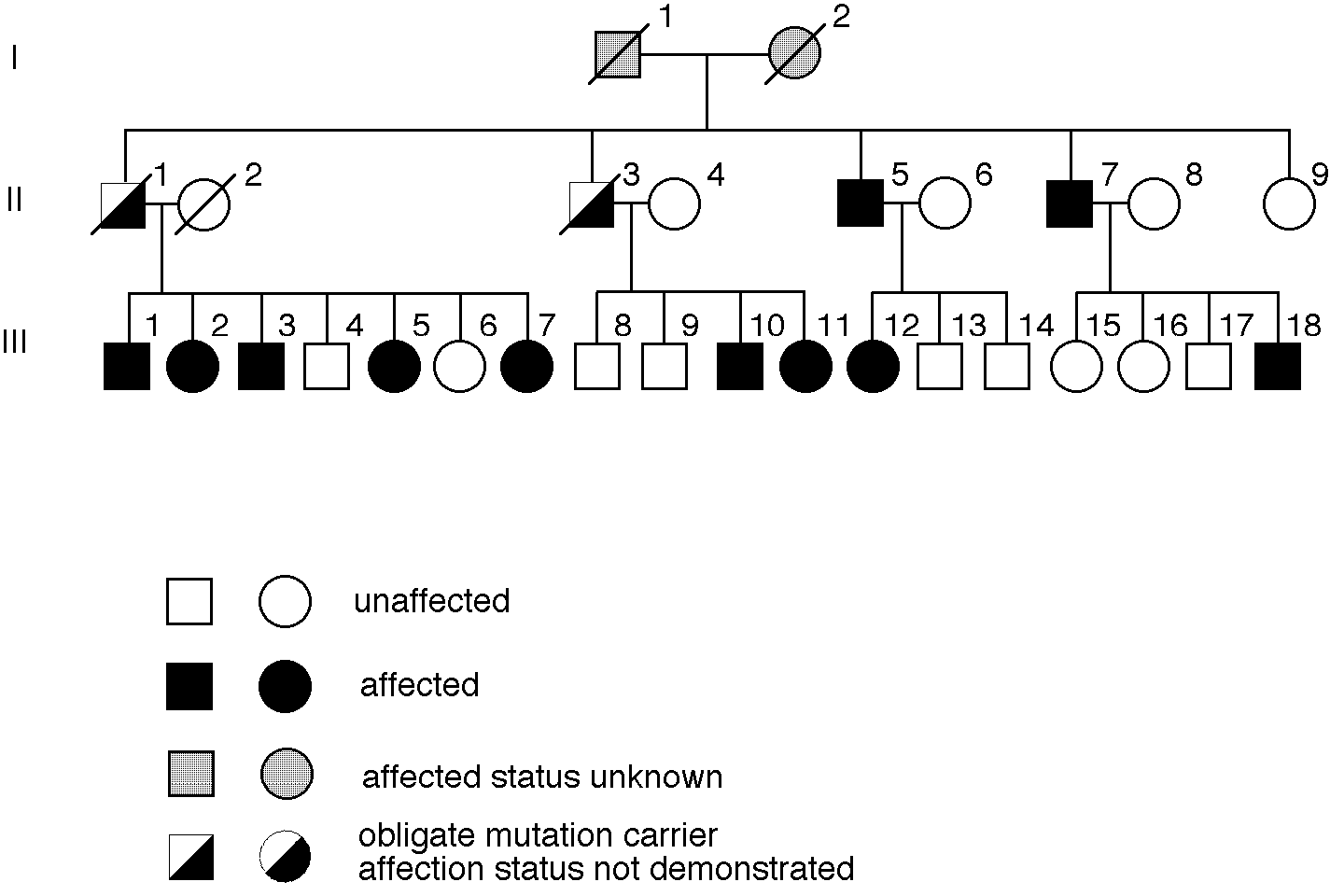
Blood was taken from 11 patients, 11 spouses, and 32 unaffected family members. Younger mutation carriers possibly were still asymptomatic. Persons under the age of 30 years were excluded from the analysis. Informed consent was obtained >from all individuals. Molecular analysis was performed in 23 probands as shown in Figure 1. Blood from one patient (III-1 in pedigree) was first obtained recently; as no further information was expected from this investigation, it was tested with only one marker.
Genomic DNA was extracted from peripheral leucocytes using standard techniques. Key individuals were typed for intragenic markers of RDS (14) and RHO (15) and for closely linked microsatellite markers D6S426 (5cM) and D6S271 (1cM) (16) on chromosome 6p and D3S1290 (3cM) and D3S1292 (3cM) (17) on chromosome 3q. Polymerase chain reaction conditions were 50-100 ng genomic DNA, 10mM Tris HCl, 50 mM KCl, 0.01% gelatin, 1.5 mM MgCl2, 200 mM of each dNTP, 10 pMol of each primer and 1 unit Taq polymerase (Stähelin) in a total volume of 25 �l. Cycling conditions were initial denaturation at 94oC for 2 minutes followed by 30 cycles of denaturation at 94oC for 30 s, annealing at 56oC for 30 s, synthesis of 1 min at 72oC followed by a final elongation at 72oC for 5 min. PCR products were visualized on a polyacrylamide gel by autoradiography or silver staining.
Linkage analysis was performed under the assumption of equal marker allele frequencies, a disease gene frequency of 0.0001 and a penetrance of 90% or 100%. Two point linkage analysis and multipoint linkage analysis were carried out using the MLINK or the LINKMAP programs of the LINKAGE package (18, 19).
RESULTS
At the RDS locus, 9 out of 10 patients shared the same allele, but segregation analysis showed that 2 patients must have inherited this allele >from the unaffected parent. The results of two point linkage analysis definitely excluded the RDS locus from being the responsible gene (see table 1), irrespective of whether penetrance was assumed to be 90% or 100%. Further support for exclusion of the RDS locus is given by the flanking microsatellites D6S426 and D6S271 (see table 1). Multipoint exclusion mapping excludes the region between markers D6S426 and D6S271, which includes the RDS gene (data not shown).
At the RHO locus, all 10 affected probands shared the same allele. Segregation analysis showed that one patient inherited this allele from his healthy parent. In addition to this result, the flanking markers D3S1290 and D3S1292 did not provide evidence for linkage to RHO. The region around D3S1290 (4 cM) and D3S1292 (13 cM) could be definitely excluded, considering a lod score of Z=-2 as a significant exclusion criteria. No positive lod score was obtained with markers RHO and D3S1292 (see table 1) assuming 100% penetrance.
Table 1: Lod scores between disease phenotype and different loci
(theta = recombination fraction, penetrance = 100%)
Markers 0.0 0.1 0.2 0.3 0.4 RDS locus RDS inf -0.413 0.018 0.108 0.054 D6S426 inf -0.731 0.085 0.220 0.096 D6S271 inf -0.900 -0.198 0.002 0.019 RHO locus RHO inf -0.729 -0.276 -0.100 -0.024 D3S1290 inf -0.509 0.022 0.133 0.065 D3S1292 inf -1.304 -0.498 -0.183 -0.044
DISCUSSION
Hereditary retinopathies including PDR and retinitis pigmentosa present a unique opportunity for studying phenomena like pleiotropy, phenotypic diversity based on allelic series, and non-allelic heterogeneity. We investigated a large Swiss family affected by PDR. The intention of this study was to exclude or confirm RDS or RHO from being involved in the pathogenesis of the disease in our family.
The results of linkage analysis definitely excludes the RDS gene from being the responsible gene in this family and thus provides evidence for genetic heterogeneity of PDR.
These data supplement at least seven previous studies that have shown RDS mutations to be responsible for fundus changes that resemble PDR. A point mutation in codon 167 and a two base pair frameshift deletion including codons 299/300 of the RDS gene have been described in PDR families (20, 21). Patients with these 2 mutations clinically presented with butterfly-shaped pigment dystrophy. Deletions of either codon 153 or 154 were shown to result in various phenotypes including retinitis pigmentosa, pattern dystrophy and fundus flavimaculatus in one family (11). A four base pair insertion in codon 140 resulted in a variable expression of fundus changes (22, 23). Remarkable clinical heterogeneity was further shown in patients with a frameshift deletion in codon 307 and with a missense mutation in codon 216 of the RDS gene (24, 25).
In a recent study of a family with eleven patients affected by PDR (21), the patients were shown to exhibit mutations in the RDS gene. Linkage analysis was performed in this family using the same intragenic microsatellite marker that we used. There was no recombination event and a positive Lod score of Z=2.4 at theta=0 was obtained in this family compared to at least one recombination event and a lod score of Z=-inf at theta=0 in our family.
We also investigated the RHO gene which is involved in a considerable proportion of patients with autosomal dominant retinitis pigmentosa (9). Despite low informativeness of the intragenic microsatellite marker we were able to exclude RHO as the disease causing locus.
Moreover, a family was recently reported in whom the clinical pictures of affected members resembled North Carolina macular dystrophy. Linkage to several loci (including the RDS gene) could be excluded. Interestingly, in three of five patients the fundus abnormalities and fluorescein angiography closely resembled those of pattern dystrophy and could thus support our findings of genetic heterogeneity (26).
We conclude that PDR can be caused by a gene or genes other than RDS. To our knowledge the study of this family provides the first evidence for genetic heterogeneity in pattern dystrophy of the retina.
ACKNOWLEDGMENTS
This work was supported by Swiss National Foundation grant 32-042 125 and in part by the Julius Klaus Foundation.
We are obliged to Prof. A. Schinzel for helpful comments on this manuscript. We thank Prof. B. H. F. Weber (Institut für Humangenetik, Julius Maximilians Universität Würzburg) for primer RDS, Dr. M. Hergersberg for support in the lab, and Mrs. Cathy Haeberlin for carefully reading the manuscript.
REFERENCES
1. Hsieh, RC; Fine, BS and Lyons JS, Patterned dystrophies of the retinal
pigment epithelium, Arch Ophthalmol 95 (1977) 429-435. ![]()
2. Marmor, MF and Byers, B, Pattern dystrophy of the pigment epithelium, Am J Ophthalmol 84 (1) (1977) 32-44.
3. de Jong, PT and Delleman, JW, Pigment epithelial pattern dystrophy, Four different manifestations in a family, Arch Ophthalmol 100 (9) (1982) 1416-1421.
4. Thomann, U; Büchi, ER; Suppiger, M; Kryenbühl, C; Schipper, I and
Spiegel, R, Age-dependent phenotypic expression of a pattern dystrophy of
the retina, Eur J Ophthalmol 5 (1995) 107-112. ![]()
5. Kajiwara, K; Hahn, LB; Mukai, S; Travis, GH; Berson, EL and Dryja, TP,
Mutations in the human retinal degeneration slow gene in autosomal dominant
retinitis pigmentosa, Nature 354 (1991) 480-483. ![]()
6. Wells, J; Wroblewski, J; Keen, J; Inglehearn, C; Jubb, C; Eckstein, A;
Jay, M; Arden, G; Bhattacharya, S; Fitzke, F and Bird, A, Mutations in the
human retinal degeneration slow (RDS) gene can cause either retinitis
pigmentosa or macular dystrophy, Nature Genet 3 (1993) 213-218. ![]()
7. Piguet, B; Heon, E; Munier, F and Stone, E, Der zunehmende Einfluss von Peripherin/RDS in der Makula-Degeneration, Ophta 4 (1995) 35.
8. Kajiwara, K; Sandberg, MA; Berson, EL and Dryja, TP, A null mutation in
the human peripherin/RDS gene in a family with autosomal dominant retinitis
punctata albescens, Nature Genet 3 (1993) 208-212. ![]()
9. Rosenfeld, PJ; McKusick, VA; Amberger, JS and Dryja, TP, Recent advances
in the gene map of inherited eye disorders: primary hereditary diseases of
the retina, choroid, and vitreous, J Med Genet 31 (1994) 903-915. ![]()
10. Travis, GH and Hepler, JE, A medley of retinal dystrophies, Nature
Genet 3 (1993) 191-192. ![]()
11. Weleber, RG; Carr, RE; Murphey, WH; Sheffield, VC and Stone, EM,
>Phenotypic variation including retinitis pigmentosa, pattern dystrophy and
fundus flavimaculatus in a single family with a deletion of codon 153 or
154 of the peripherin/RDS gene, Arch Ophthalmol 111 (1993) 1531-1542. ![]()
12. Shastry, BS, Retinitis pigmentosa and related disorders: phenotypes of
rhodopsin and peripherin /RDS mutations, Am J Med Genet 52 (1994) 467-474. ![]()
13. Bird, AC, Retinal photoreceptor dystrophies, Am J Ophthalmol 119 (1995)
543-562. ![]()
14. Kumar-Singh, R; Jordan, SA; Farrar, GJ and Humphries, P, Poly (T/A)
polymorphism at the human retinal degeneration slow (RDS) locus, Nucl
Acids Res 19 (20) (1991) 5800. ![]()
15. Weber, JL and May, PE, Abundant class of human DNA polymorphisms which
can be typed using the polymerase chain reaction, Am J Hum Genet 44 (3)
(1989) 388-396. ![]()
16. Gyapay, G; Morissette, J; Vignal, A; Dib, C; Fizames, C; Millasseau, P;
Marc, S; Bernardi, G; Lathrop, M and Weissenbach, J, The 1993-94 Généthon
human genetic linkage map, Nature Genet 7 (1994) 246-339. ![]()
17. Weissenbach, J; Guapay, G; Dib, C; Vignal, A; Morissette, J;
Millasseau, P; Vaysseix, G and Lathrop, M, A second-generation linkage map
of the human genome, Nature 359 (1992) 794-801. ![]()
18. Lathrop, GM; Lalouel, JM; Julier, C and Ott, J, Strategies for
multipoint linkage analysis in humans, Proc. Natn. Acad. Sci. U.S.A. 81
(1984) 3443-3446. ![]()
19. Ott, J, Analysis of human genetic linkage. Revised edn. John Hopkins University Press, Baltimore London (1991).
20. Nichols, BE; Drack, AV; Vandenburgh, K; Kimura, AE; Sheffield, VC and
Stone, EM, A 2 base pair deletion in the RDS gene associated with
butterfly-shaped pigment dystrophy of the fovea, Hum Mol Genet 2 (5) (1993)
601-603. ![]()
21. Nichols, BE; Sheffield, VC; Vandenburgh, K; Drack, AV; Kimura, AE and
Stone, EM, Butterfly-shaped pigment dystrophy of the fovea caused by a
point mutation in codon 167 of the RDS gene, Nature Genet 3 (1993) 202-207. ![]()
22. Keen, TJ; Inglehearn, CF; Kim, R; Bird, AC and Bhattacharya, S, Retinal
pattern dystrophy associated with a 4 bp insertion at codon 140 in the
RDS-peripherin gene, Hum Mol Genet 3 (2) (1994) 367-368. ![]()
23. Kim, RY; Dollfus, H; Keen, TJ; Fitzke, FW; Arden, GB; Bhattacharya, SS
and Bird, AC, Autosomal dominant pattern dystrophy of the retina associated
with a 4-base pair insertion at codon 140 in the peripherin/rds gene, Arch
Ophthalmol 113 (1995) 451-455. ![]()
24. Apfelstedt-Sylla, E; Theischen, M; Ruther, K; Wedemann, H; Gal, A and
Zrenner, E, Extensive intrafamilial and interfamilial phenotypic variation
among patients with autosomal dominant retinal dystrophy and mutations in
the human RDS/peripherin gene, Br J Ophthalmol 79 (1) (1995) 28-34. ![]()
25. Richards, SC and Creel, DJ, Pattern dystrophy and retinitis pigmentosa
caused by a peripherin/RDS mutation, Retina 15 (1) (1995) 68-72. ![]()
26. Holz, FG; Evans, K; Gregory, CY; Bhattacharya, S and Bird, AC,
Autosomal dominant macular dystrophy simulating North Carolina macular
dystrophy, Arch Ophthalmol 113 (2) (1995) 178-184. ![]()